
Rozwój choroby Alzheimera tłumaczy się w oparciu o dwie hipotezy. Pierwszą z nich jest hipoteza cholinergiczna. Jest ona oparta na występowaniu zaburzeń układu cholinergicznego, przekładających się na nieprawidłowe funkcjonowanie neuroprzekaźnictwa cholinergicznego, związanego przede wszystkim z acetylocholiną, która odgrywa główną rolę w procesach uczenia się i pamięci. Hipoteza ta zyskała na znaczeniu, ponieważ współczesna terapia opiera się jedynie na lekach będących inhibitorami cholinoesteraz, enzymów odpowiedzialnych za rozkład acetylocholiny. Druga hipoteza – kaskady β-amyloidu (Aβ) zakłada, że istotne znaczenie w patogenezie choroby Alzheimera stanowi odkładanie się złogów Aβ w postaci blaszek amyloidowych w korze mózgowej chorych. Pozostałe zmiany neuropatologiczne: zwyrodnienie włókienkowe neuronów typu Alzheimera (NFTs), zwyrodnienie synaps oraz zanik neuronów mają charakter wtórny i są następstwem gromadzenia się Aβ.
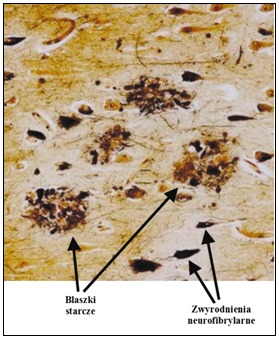
Rys.1 Blaszki starcze (ang. senile plaques) i zwyrodnienia neurofibrylarne w korze mózgowej chorego na chorobę Alzheimera (Blennow i in., 2006).
Blaszki starcze (ang. senile plaques) są to złogi Aβ, przyjmujące patologiczną konformację w formie struktury β. Aβ jest jedynie fragmentem (40-43-aminokwasowym) znacznie dłuższego białka APP (398-770 aminokwasów). Potwierdzeniem są następujące dowody: powiązanie wystąpienia choroby Alzheimera z dziedziczonymi mutacjami APP i z nadekspresją APP u chorych na zespół Downa oraz neurotoksyczność fibryli Aβ. Stwierdzono już ok. 16 mutacji APP, ale uznaje się, że są one przyczyną nie więcej niż 1,5% wszystkich przypadków zachorowań.
Prawidłowe białko APP jest transmembranową glikoproteiną, która występuje według jednych źródeł w trzech izoformach, a według innych aż w sześciu. Powstają one w wyniku alternatywnego splicingu mRNA. APP jest normalnie rozszczepiana w obrębie domeny Aβ, w skutek czego powstaje forma wydzielnicza (ang. secreted – Aβs). W alternatywnym szlaku APP ulega rozszczepieniu w innych miejscach: domenie pozakomórkowej i domenie transmembranowej, co daje rozpuszczalny Aβ, uważany za potencjalne źródło płytek amyloidowych w chorobie Alzheimera.
Jednym z pierwszych zaproponowanych szlaków metabolicznych APP było rozszczepienie normalnego Aβ pomiędzy 16. a 17. resztą aminokwasową przez proteazę zwaną α-sekretazą. Uwalnia ona duże rozpuszczalne białko zawierające tylko sekwencję N-końcową Aβ. Natomiast dwie inne sekretazy, β- i γ-sekretaza przecinają wiązania pomiędzy aminokwasami, odpowiednio na N- i C-końcu Aβ. W rezultacie wzrasta ilość zarówno normalnego wydzielniczego amyloidu Aβs, jak i amyloidu 40-42-aminokwasowego występującego w depozytach amyloidowych. Warto tutaj wspomnieć o roli presenilin, szczególnie PS1. Sugeruje się bowiem, że albo pełni ona funkcję kofaktora dla γ-sekretazy, albo sama jest tym enzymem.
Im Aβ jest dłuższy, tym wykazuje większą zdolność do agregacji. Forma Aβ1-42 zawiera dwie dodatkowe hydrofobowe reszty aminokwasowe, przez co łatwiej fibrylizuje i agreguje. Ponadto stwierdzono, że zachodząca w trakcie amyloidogenezy zmiana konformacji w amyloidzie β z formy α-helisy na β-kartkę warunkuje jego nierozpuszczalność i neurotoksyczność, a także odporność na działanie proteaz. Gromadzenie Aβ mogą też wzmagać pewne czynniki, które się z nim wiążą. Należą do nich: apolipoproteina ApoE, proteoglikany siarczanu heparyny oraz metale ciężkie: cynk i aluminium. Ponadto agregację może także wywoływać stres oksydacyjny.
Przeprowadzone analizy z wykorzystaniem mikroskopii elektronowej wykazały, że powstające fibryle amyloidowe są prostymi, nierozgałęzionymi włóknami o średnicy 70-120 Å i nieokreślonej długości. Fibryle amyloidowe są odkładane w płytkach starczych rozmieszczonych pozakomórkowo w korze mózgu u chorych na Alzheimera. Obecnie wyróżnia się dwa rodzaje złogów Aβ. Jest to depozyt ogniskowy – blaszka neurytyczna (ang. neuritic plaque) i rozproszony (ang. diffuse). Blaszki oprócz Aβ zawierają także inne białka tzw. towarzyszące, m. in. ApoE i ApoJ, białko tau, α-synukleinę, ubikwitynę i in., a także ułożony na zewnątrz wieniec komórek astroglejowych i komórki mikrogleju, rozmieszczone wewnątrz złogu między wieńcem i rdzeniem lub w samym centrum blaszki.
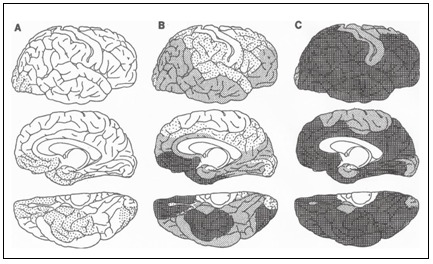
Rys.2 Schemat rozmieszczenia depozytów amyloidu β w kolejnych stadiach rozwoju choroby Alzheimera (Braak, Braak, 1991).
Strategie terapeutyczne
Terapie stosowane na dzień dzisiejszy poprawiają pamięć, łagodzą problemy behawioralne i jakość życia osób dotkniętych chorobą Alzheimera, ale nie wywierają znaczącego wpływu na postęp choroby. W oparciu o cholinergiczną hipotezę rozwoju choroby Alzheimera oraz zgodnie z postanowieniami AAN ( American Academy of Neurology), FDA (Food and Drug Administration) i NICE (UK National Institute of Clinical Excellence), za najskuteczniejszą metodę leczenia choroby Alzheimera uznano inhibitory esteraz cholinowych. Hamują one aktywność tych enzymów w celu zmniejszenia bądź zahamowania degradacji acetylocholiny. W podobnym ujęciu rozważane są także inne leki wywierające wpływ na neuroprzekaźnictwo cholinergiczne, a mianowicie agoniści receptorów acetylocholiny, a także leki o działaniu dopaminergicznym i noradrenergicznym, leki serotoninergiczne i gabaergiczne oraz aminokwasy pobudzające. Dla większości z nich udowodniono działanie prokognitywne, a mimo to brak wiarygodnych danych, by rekomendować je do terapii choroby Alzheimera. W odniesieniu do niektórych są niezbędne dalsze badania kliniczne.
Poza lekami wpływającymi na neuroprzekaźnictwo nadal poszukiwane są nowe strategie terapeutyczne, takie jak: leki nootropowe, neurotrofiny, estrogeny, środki zapobiegające syntezie białka tau i gromadzeniu Aβ, leki przeciwzapalne, psychostymulujące, poprawiające krążenie mózgowe i obniżające ciśnienie, przeciwzakrzepowe, obniżające hipoksję ośrodkowego układu nerwowego, środki chelatujące glin, antagoniści kanałów wapniowych oraz neuropeptydy. Badania zmierzają nie tylko do łagodzenia objawów rozwijającej się choroby, ale także podejmowane są próby ochrony przed wystąpieniem choroby Alzheimera w grupie o podwyższonym ryzyku zachorowania. Stąd też coraz większym zainteresowaniem cieszą się badania nad tzw. szczepionką przeciw chorobie Alzheimera.
Immunoterapia
Szczepienie przeciwko chorobie Alzheimera wprowadził jako pierwszy zespół Dale’a Schenka w 1999 roku. Używając Aβ zimmunizowali oni młode myszy transgeniczne o zmutowanym białku prekursorowym amyloidu ludzkiego APP (myszy APP tg). W trakcie doświadczenia zauważono znaczącą redukcję ilości depozytów amyloidowych w porównaniu do grupy nieimmunizowanych myszy APP tg. Ponadto odkryto, że podanie Aβ starszym myszom APP tg, u których depozyty amyloidu zaczęły się już odkładać, zaowocowało redukcją płytek starczych i chroniło przed dalszym odkładaniem się amyloidu. Podobne efekty uzyskano po podaniu przeciwciał monoklonalnych lub poliklonalnych w stosunku do Aβ. Pierwsze wzmianki o użyciu przeciwciał dla Aβ jako narzędzia terapeutycznego przedstawił zespół Solomona. Zaprezentował on badania, w których monoklonalne przeciwciała anty-Aβ zapobiegały w warunkach in vitro tworzeniu włókien Aβ, a poza tym sprzyjały rozpuszczaniu wstępnie uformowanych fibryli amyloidowych.
Badania kliniczne szczepionki Aβ u pacjentów cierpiących na chorobę Alzheimera
W 2000 roku rozpoczęto I fazę badań klinicznych nad szczepionką AN-1792 składającą się z syntetycznego Aβ1-42 i adjuwantu immunologicznego QS21. We wrześniu 2001 roku rozpoczęto drugą fazę badań klinicznych w grupie chorych znajdujących się w średnim stadium choroby. Jednak po 6 miesiącach badania zostały wstrzymane. Powodem było wystąpienie u 6% badanych efektów ubocznych, spowodowanych prawdopodobnie dodatkiem Polisorbatu-80. Mimo to udało się stwierdzić redukcję liczby płytek starczych oraz przesiąkanie fagocytów zawierających Aβ, co sugeruje oczyszczanie depozytów amyloidu po immunizacji pacjentów za pomocą Aβ. Zauważono również zanik skojarzonych z płytkami neurytów dystroficznych zawierających ufosforylowane białko tau. Bez zmian pozostał amyloid naczyniowy oraz zwyrodnienia włókienkowe neuronów NFTs. Jednakże przy znaczącej redukcji płytek nie udało się stwierdzić jednoznacznej poprawy funkcji poznawczych. Na podstawie licznych przesłanek stwierdzono również, że szczepionka AN-1792 powoduje aktywację Aβ-reaktywnych komórek Th1, subpopulacji limfocytów T.
Szczepionki przeciwko Aβ
1. Szczepionki peptydowe
a. N-końcowy fragment Aβ z nie powodującym zapalenia mózgu epitopem limfocytu T oraz białkiem nośnikowym.
Jest to krótki N-koniec Aβ1-15, który jest połączony z epitopem limfocytu T pochodzącym z surowiczej albuminy wołowej. Aby wzmocnić odpowiedź przeciwciał używa się peptydów wielo-antygenowych. W tym celu do końca peptydu dołącza się reszty lizyny. W podobny sposób można wzmocnić odpowiedź komórek Th2, aby uniknąć wywołania zapalenia mózgu powodowanego często przez Th1. Osiąga się to przez przyłączanie np.: HLA-DR (cząsteczkę II klasy układu zgodności tkankowej człowieka) lub mannanu, który wzmacnia wychwyt antygenów przez komórki dendrytyczne i produkcję IgG1, IL-10 czy IL-4. Wzmocnienie odpowiedzi w postaci komórek Th2 jest również możliwe poprzez immunizacje donosową.
b. Oparta na liposomach terapeutyczna szczepionka przeciwko Aβ
Szczepionka taka powstaje w skutek przyłączenia do obydwu końców peptydowego fragmentu Aβ1-15 16-węglowego kwasu palmitynowego. Drugim sposobem jest dodanie do obydwu końców Aβ1-15 glikolu polietylenowego (PEG). Pierwsza z nich rozpoznaje strukturę β kartki Aβ, a druga – α helissy.
c. Szczepionka wchłaniana przez skórę
Zespół Nikolica podał taką szczepionkę ogolonym myszom. Był to roztwór zawierający peptyd Aβ1-42 oraz toksynę cholery. Szczepionka taka ma za zadanie indukować odpowiedź immunologiczną Th2, którą promują zawarte w naskórku komórki Langerhansa z receptorami CD14.
d. Cząsteczki wirusa mysiej białaczki wykazujące ekspresję peptydu Aβ1-15 i receptora PDGF
Wiruso-podobne cząsteczki wykazujące ekspresję Aβ1-15 i receptora PDGF są produkowane prze komórki HEK-293 (komórki nerek ludzkich embrionów) po uprzedniej ich kotransfekcji plazmidami: pHIT60 zawierającym geny gag i pol wirusa mysiej białaczki oraz pDisplay zawierającym peptyd Aβ1-15, receptor PDGF oraz sekwencję sygnałową Igκ. Uzyskane białko fuzyjne podawano dożylnie myszom szczepu APP23 (z mutacją APP pod kontrolą promotora Thy1; płytki amyloidowe powstają w ciągu 6 miesięcy, a w naczyniach krwionośnych mózgu występuje Aβ39-41). Charakteryzuje się on prawie 7-krotną nadekspresją APP, obecnością rozpuszczalnych płytek w korze nowej i hipokampie, astrocytozą oraz mikroglejozą. Wykazały one wysokie miano wyprodukowanych przeciwciał anty-Aβ, a dokładniej IgG1 i IgG2b. Ponadto stwierdzono znaczącą redukcję liczby płytek starczych, a także rozpuszczalnych i nierozpuszczalnych form Aβ.
e. Szczepionka ACC-001
Jest to N-końcowy fragment peptydu Aβ sprzężony z toksyną błoniczą (wytwarzaną przez pałeczki Corynebacterium diphteriae) i QS21 jako adjuwantem. Zastosowano N-koniec Aβ, aby wyeliminować ryzyko rozwoju zapalenia mózgu warunkowane przez epitop zlokalizowany na C-końcu. Jednakże nie ma pewności, że u ludzi wszystkich ras epitop ten zlokalizowany jest tylko i wyłącznie na C-końcu Aβ. Ponadto rozważa się również problem zastosowania QS21, który wzmaga aktywność Th1, przez co przyczynia się do rozwoju stanu zapalnego w centralnym układzie nerwowym.
f. szczepionka AD02 Jest to krótki amino-końcowy fragment Aβ (Aβ1-6), będący pochodną N-końca Aβ stanowiącego epitop dla limfocytów B. Dzięki temu unika się aktywacji limfocytów T. Tak powstały pełnowartościowy antygen pozwala rozwiać wątpliwości dotyczące bezpieczeństwa szczepionki AN1792 (Aβ1-42), posiadającej pozostałości Aβ15-42. Fragment ten stanowi najczęściej epitop dla limfocytów T, przez co prawdopodobnie przyczynia się do aktywacji limfocytów Th1, a co za tym idzie jest odpowiedzialny za autoimmunologiczne zapalenie mózgu obejmujące opony mózgowe. Głównym celem szczepionki AD02 ma być zapobieganie budowaniu złogów amyloidowych w mózgu. Dokonuje się to poprzez wytworzenie dodatkowych przeciwciał i zaatakowanie tej części amyloidu, który jest odpowiedzialny za powstawanie złogów. Mówimy tutaj zatem o szczepionce terapeutycznej, gdyż atakuje ona już istniejący stan chorobowy. Schneeberger przeprowadził przedkliniczne testy szczepionki AD02 na myszach i wykazał, że dochodzi do indukcji odpowiedzi immunologicznej przeciwko Aβ w postaci zagregowanej, ale nie przeciwko APP. Stwierdził także jej skuteczność w redukcji zmian chorobowych astrocytów oraz akumulacji mikrogleju.
2. Szczepionka DNA
Szczepionka DNA jest tworzona poprzez wprowadzenie do wybranego plazmidu cDNA jednego, dwóch lub kilku fragmentów Aβ1-42, sekwencji promotora, sekwencji liderowej (warunkującej właściwe umiejscowienie rybosomu na mRNA) oraz sekwencji kierującej (decydującej o przyszłej lokalizacji syntetyzowanych białek). Przykładem takiej szczepionki jest rozwiązanie zaproponowane przez zespół Qu. Wykorzystali oni w tym celu promotor SP72 (syntetyczny promotor specyficzny dla komórek ssaków), sekwencję liderową α-antytrypsyny oraz sekwencję kierującą do białek MHC klasy II. Utworzona cząsteczka testowana była na myszach, którym podawano ją śródskórnie w uszy. Zaobserwowano zadowalającą odpowiedź immunologiczną względem Aβ bez jednoczesnej aktywacji cytotoksycznych limfocytów T. W kolejnych etapach badań ten sam zespół eksperymentował z innymi sekwencjami liderowymi (Adenowirusa E3) czy wprowadzeniem sekwencji aktywatora transkrypcji GAL4 z jednoczesną zmianą promotora na CMV (promotor tetracyklinowy wirusa cytomegalii). Zaobserwowano znaczący wzrost liczby przeciwciał typu Th2 oraz redukcję Aβ. Z kolei w wyniku porównania szczepionki DNA ze szczepionką peptydową Qu i jego współpracownicy wysunęli wstępny wniosek, że ta pierwsza zdecydowanie indukuje odpowiedź immunologiczną z udziałem Th2.
Zespół Okury stworzył podobną szczepionkę DNA, ale z wykorzystaniem plazmidu pTarget, sekwencji sygnałowej Igκ oraz Aβ1-42. Kilkukrotnie powtórzone domięśniowe podanie tej szczepionki myszom APP tg zaowocowało redukcją Aβ bez aktywacji limfocytów T.
Twórcami kolejnej szczepionki DNA był zespół Movsesyana. Stanowi ją plazmid pCMV z sekwencją sygnalną chemokiny IP10, genem kodującym chemokinę CCL22, 3 powtórzenia Aβ1-11 oraz gen PADRE (kodujący sekwencję aminokwasów: Pro-Ala-Asp-Arg-Glu). W eksperymencie wykorzystano mutanty myszy APP tg x PS1 tg x tau tg (3x-Tg-AD), którym podawano szczepionkę 3-krotnie w ogoloną skórę brzucha z użyciem pistoletu genowego. Użycie CCL22 spowodowało odpowiedź immunologiczną z udziałem Th2 i produkcję przeciwciał klasy IgG1 i IgG2b. Wiadomo też, że liczba płytek starczych uległa znacznej redukcji oraz nastąpiła poprawa funkcji poznawczych. Dodatkowo nierozpuszczalne amyloidy: Aβ40 i Aβ42 oraz rozpuszczalne oligomery Aβ (trimery i heksamery) uległy znaczącej redukcji, jednakże nie nastąpiła żadna oczekiwana zmiana w patologii białka tau.
Szczepionka zawierająca Aβ1-42 oraz zmutowany gen kaspazy charakteryzuje się tym, że nie wywołuje apoptozy, ale wystarcza by wzbudzić apoptosomy, co pociąga za sobą zwiększoną prezentację antygenów. Zespół DaSilvy zainteresował się tym mechanizmem, ponieważ post-mitotyczne komórki mięśniowe charakteryzują się bardzo ubogą prezentacją antygenową. Powstałą szczepionkę podano domięśniowo myszom TGCRND8 (myszy z wielokrotnymi mutacjami genu APP, u których płytki amyloidowe powstają wciągu 3 miesięcy). Wykazały one odpowiedź przeciwciał typu Th2 oraz redukcję płytek amyloidowych i nierozpuszczalnego Aβ42. Zauważono również nieznaczną redukcję mózgowej angiopatii amyloidowej oraz rozpuszczalnych oligomerów Aβ.
3. Rekombinanty
W 2005 roku uzyskano zrekombinowany ziemniak wykazujący ekspresję tandemowo ułożonych pięciu fragmentów Aβ1-42. Uzyskany z niego ekstrakt białkowy charakteryzował się zawartością 0,9 µg Aβ w 1 mg ekstraktu białkowego. Myszom Tg2576 (z mutacją APP pod kontrolą prionowego promotora chomika; rozwój płytek amyloidowych następuje w ciągu 9 miesięcy) podawano ten ekstrakt w ilości 25 mg łącznie z podjednostką B toksyny cholery (CTB) pełniącą rolę adjuwantu. Po 3 tygodniach zauważono podwyższony poziom przeciwciał anty-Aβ i redukcję płytek starczych. Na podstawie tych obserwacji zasugerowano, że w pewnych warunkach doustne podawanie przygotowanego tak ekstraktu łącznie z adjuwantem może przełamać tolerancję immunologiczną. Jednak już wkrótce pojawił się problem – co się stanie, gdy ziemniaki zostaną ugotowane? Dlatego też postanowiono zrekombinować takie warzywo, które można spożywać bez potrzeby przeprowadzenia obróbki cieplnej. Wybór padł na pomidora. Jednak nie stwierdzono produkcji przeciwciał anty-Aβ. Sytuacja uległa zmianie dopiero wówczas, gdy myszom tuż po karmieniu wstrzyknięto niewielką ilość Aβ, co sugeruje, że nabyły one odporność poprzez zjedzenie pomidora zawierającego Aβ.
Podobne doświadczenia żywieniowe przeprowadzono z wykorzystaniem jako „nośnika” dla Aβ zrekombinowanych komórek niepatogenicznego szczepu Salmonelli. Zespół Boutajangouta zaobserwował redukcję Aβ. Jednakże wydzielany przez komórki bakterii Aβ nie mógł być immunogenny. Faktycznie wydaje się, że immunogenność zachodzi dopiero wówczas, gdy komórki bakterii zostają sfagocytowane przez komórki M (komórki nabłkonkowe związane z grudkami) lub komórki dendrytyczne.
Inną procedurę badawczą stanowi wykorzystanie fagów. Frenkel wraz z współpracownikami stworzyli zrekombinowanego faga, który charakteryzował się ekspresją 10 kopii Aβ3-6 w obrębie białka III (pIII) lub 300 kopii w obrębie głównego białka osłonki (pVIII). Przygotowane inokulum podano kilkakrotnie myszom APP tg dootrzewnowo lub donosowo. Spowodowało to produkcję przeciwciał dla Aβ, znaczącą redukcje płytek starczych oraz poprawę rezultatów z labiryntu wodnego Morrisa. Uzyskane przeciwciała zapobiegały agregacji Aβ i powodowanej przez niego neurotoksyczności.
4. Zrekombinowane wektory wirusowe
W tworzeniu szczepionki kierowanej przeciwko Aβ sięgnięto również po rekombinowane wektory wirusowe. Przykłady tych wektorów umieszczono w Tabeli 1. Zawierają one sekwencje Aβ, choć różnią się długością wbudowywanych fragmentów peptydowych lub liczbą ich powtórzeń.
Tab.1 Przykłady konstrukcji rekombinowanych wektorów wirusowych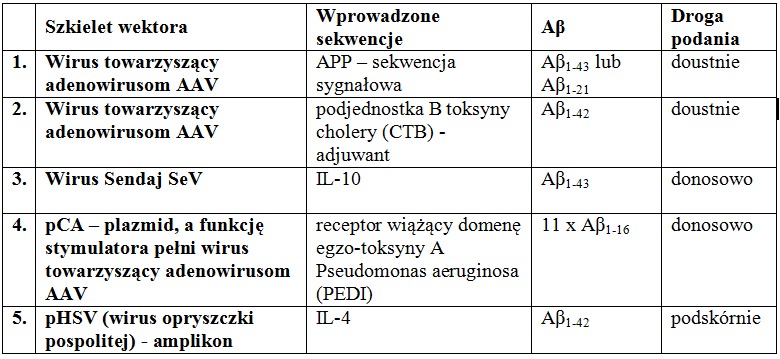
U zaszczepionych myszy zaobserwowano podwyższone stężenie przeciwciał anty-Aβ. Należaly do nich zdecydowanie IgG1 i IgG2b. Znaczącej redukcji uległa również liczba płytek starczych. W celu oceny pamięci przestrzennej i funkcji poznawczych myszy poddano licznym testom pamięci, m.in.: testowi Y-labiryntu, testowi wodnego labiryntu Morrisa, testowi rozpoznawania nowego obiektu oraz testowi warunkowania strachu. Stwierdzono zasadniczą poprawę uzyskanych wyników, a przebadane następnie mózgowia charakteryzowały się redukcją płytek starczych, rozpuszczalnych fragmentów Aβ40 i Aβ42, oraz rozpuszczalnych nona- i dodekamerów. Dalsze badania na małpach potwierdziły redukcję płytek starczych bez występowania niepożądanych efektów ubocznych. Wydaje się również, że w odpowiedzi immunologicznej główną rolę odegrały limfocyty Th2, a zatem wyeliminowane zostało ryzyko wystąpienia zapalenia mózgu warunkowanego przez Th1. Może się jednak zdarzyć, że produkty niektórych wprowadzanych genów mogą prowadzić do odpowiedzi immunologicznej z udziałem Th1. Ponadto w zależności od szczepionki, w mniejszych lub większych ilościach, obok IgG1 i IgG2b pojawiają się także inne przeciwciała. Chociaż wydaje się, że zastosowane wektory są bezpieczne dla organizmu człowieka, większość wprowadzonych genów jest szybko usuwana lub episomy zostają zatrzymane, a komórki nabłonka jelitowego ulegają szybkiej odnowie, to jednak pojawia się ryzyko i konieczne jest przeprowadzenie dalszych badań.
Kamila Borowiec, Sylwia Kowalczyk
Literatura:
1. Agadjanyan M.G., Ghochikyan A., Petrushina I., Vasilevko V., Movsesyan N., Mkrtichyan M., Saing T., Cribbs D.H., 2005. Prototype Alzheimer’s disease vaccine using the immunodominant B cell epitope from β-amyloid and promiscuous T cell epitope pan HLA DR-binding peptide. Journal of Immunology 174, 1580 – 1586.
2. Barcikowska M., Bilikiewicz A., 2004. Choroba Alzheimera w teorii i praktyce klinicznej, Wydawnictwo Czelej Sp. z o. o., Lublin.
3. Blennow K., de Leon M. J., Zetterberg H., 2006. Alzheimer’s disease. Lancet 368, 387 – 397.
4. Boutajangout A., Goni F., Knudsen E., Schreiber F., Asuni A., Quartermain D., Frangione B., Chabalgoity A., Wisniewski T., Sigurdsson E.M., 2009. Diminished amyloid-β burden in Tg2576 Mice following a prophylactic oral immunization with a salmonella-based amyloid-β derivative vaccine. Journal of Alzheimer’s Disease 18, 961 – 972.
5. Braak H., Braak E., 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica 82, 239 – 257.
6. Castellani R. J., Lee H.-G., Zhu X., Nunomura A., Perry G., Smith M. A., 2006. Neuropathology of Alzheimer disease: pathognomonic but not pathogenic. Acta Neuropathologica 111, 503 – 507.
7. Contestabile A., 2011. The history of the cholinergic hypothesis. Behavioural Brain Research 221, 334 – 340.
8. DaSilva K.A., Brown M.E., McLaurin J., 2009. Reduced oligomeric and vascular amyloid-β following immunization of TgCRND8 mice with an Alzheimer’s DNA vaccine. Vaccine 27, 1365 – 1376.
9. Frazer M.E., Hughes J.E., Mastrangelo M.A., Tibbens J.L., Federoff H.J., Bowers W.J., 2008. Reduced pathology and improved behavioral performance in Alzheimer’s disease mice vaccinated with HSV amplicons expressing amyloid-β and interleukin-4. Molecular Therapy 16, 845 – 853.
10. Ghochikyan A., Petrushina I., Lees A., Vasilevko V., Movsesyan N., Karapetyan A., Agadjanyan M.G., Cribbs D.H., 2006. Aβ-immunotherapy for Alzheimer’s disease using mannan-amyloid-beta peptide immunoconjugates. DNA and Cell Biology 25, 571 – 580.
11. Goedert M., Spillantini M. G., 2006. A century of Alzheimer’s disease. Science 314, 777 – 778.
12. Hara H., Monsonego A., Yuasa K., Adachi K., Xiao X., Takeda S., Takahashi K., Weiner H. L., Tabira T., 2004. Development of a safe oral Aβ vaccine using recombinant adeno-associated virus vector for Alzheimer’s disease. Journal of Alzheimer’s Disease 6, 483 – 488.
13. Hardy J., Selkoe D. J., 2002. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297, 353 – 356.
14. Kubis A. M., Janusz M., 2008. Choroba Alzheimera – nowe możliwości terapeutyczne oraz stosowane modele eksperymentalne. Postępy Higieny i Medycyny Doświadczalnej 62, 372 – 392.
15. Leszek J., 1998. Choroba Alzheimera. Praca zbiorowa pod redakcją J. Leszka, Wydawnictwo Volumed, Wrocław.
16. Morgan D., Diamond D.M., Gottschall P.E., Ugen K.E., Dickey C., Hardy J., Duff K., Jantzen P., DiCarlo G., Wilcock D., Connor K., Hatcher J., Hope C., Gordon M., Arendash G.W., 2000. Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 408, 982 – 985.
17. Morgan D., Gitter B. D., 2004. Evidence supporting a role for anti-Aβ antibodies in the treatment of Alzheimer’s disease. Neurobiology of Aging 25, 605 – 608.
18. Movsesyan N., Ghochikyan A., Mkrtichyan M., Petrushina I., Davtyan H., Olkhanud P.B., Head E., Biragyn A., Cribbs D. H., Agadjanyan M.G., 2008. Reducing AD-like pathology in 3xTg-AD mouse model by DNA epitope vaccine – A novel immunotherapeutic strategy. PLoS ONE 3, art. no. e2124.
19. Muhs A., Hickman D.T., Pihlgren M., Chuard N., Giriens V., Meerschman C., Van Der Auwera I., van Leuven I., Sugawara M., Weingertner M. C., Bechinger B., Greferath R., Kolonko N., Nagel-Steger L., Riesner D., Brady R. O., Pfeifer A., Nicolau C., 2007. Liposomal vaccines with conformation-specific amyloid peptide antigens define immune response and efficacy in APP transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 104, 9810 – 9815.
20. Nicolll J.A.R., Wilkinson D., Holmes C., Steart P., Markham, H., Weller R.O., 2003. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: A case report. Nature Medicine 9, 448 – 452.
21. Nikolic W.V., Bai Y., Obregon D., Hou H., Mori T., Zeng J., Ehrhart J., Shytle R. D., Giunta B., Morgan D., Town T., Tan J., 2007. Transcutaneous β-amyloid immunization reduces cerebral β-amyloid deposits without T cell infiltration and microhemorrhage. Proceedings of the National Academy of Sciences of the United States of America 104, 2507 – 2512.
22. Okura Y., Miyakoshi A., Kohyama K., Park I.-K., Staufenbiel M., Matsumoto Y., 2006. Nonviral Aβ DNA vaccine therapy against Alzheimer’s disease: Long-term effects and safety. Proceedings of the National Academy of Sciences of the United States of America 103, 9619 – 9624.
23. Perihar M. S., Hemnani T., 2004. Alzheimer’s disease pathogenesis and therapeutic interventions. Journal of Clinical Neuroscience 11, 456 – 460.
24. Qu B., Rosenberg R.N., Li L., Boyer P.J., Johnston S.A., 2004. Gene vaccination to bias the immune response to amyloid-β peptide as therapy for Alzheimer disease. Archives of Neurology 61, 1859 – 1864.
25. Qu B.-X., Xiang Q., Li L., Johnston S.A., Hynan L.S., Rosenberg R.N., 2007. Aβ42 gene vaccine prevents Aβ42 deposition in brain of double transgenic mice. Journal of the Neurological Sciences 260, 204-213.
26. Schenk D., Barbour R., Dunn W., Gordon G., Grajeda H., Guldo T., Hu K., Huang J., Johnson-Wood K., Khan K., Kholodenko D., Lee M., Liao Z., Lieberburg I., Motter R., Mutter L., Soriano F., Shopp G., Vasquez N., Vaandevert C., Walker S., Wogulis M., Yednock T., Games D., Seubert P., 1999. Immunization with amyloid-β attenuates Alzheimer disease-like pathology in the PDAPP mouse. Nature 400, 173 – 177.
27. Schneeberger A., Mandler M., Otava O., Zauner W., Mattner F., Schmidt W., 2009. Development of AFFITOPE vaccines for Alzheimer’s Disease (AD) – From concept to clinical testing. Journal of Nutrition, Health and Aging 13, 264 – 267.
28. Serpell L. C., 2000. Alzheimer’s amyloid fibrils: structure and assembly. Biochemica et Biophysica Acta 1502, 16 – 26.
29. Tabira T., 2010. Immunization therapy for Alzheimer disease: a comprehensive review of active immunization strategies. Tohoku Journal of Experimental Medicine 220, 95 – 106.
30. Wisniewski T., Ghiso J., Frangione B., 1997. Biology of Aβ amyloid in Alzheimer’s disease. Neurobiology of Disease 4, 313 – 328.
31. Yankner B. A., 1996. Mechanism of neuronal degeneration in Alzheimer’s disease. Neuron 16, 921 – 924.
32. Zhang J., Wu X., Qin C., Qi J., Ma S., Zhang H., Kong Q., Chen D., Ba D., He W., 2003. A novel recombinant adeno-associated virus vaccine reduces behavioral impairment and β-amyloid plaques in a mouse model of Alzheimer’s disease. Neurobiology of Disease 14, 365 – 379.