RZEDRUK, oryginał dostępny pod adresem www
Politechnika Śląska (www)
Wydział Chemiczny (www)
Katedra Chemii Organicznej, Bioorganicznej i Biotechnologii (www)
Kierownik Katedry: dr hab. inż. Mirosław Gibas prof. Politechniki Śląskiej
Adres:
ul. B. Krzywoustego 4
44-100 Gliwice
Kinetyka reakcji enzymatycznych
(na podstawie „Ćwiczenia z biochemii”, praca zbiorowa pod redakcją Leokadii Kłyszejko-Stefanowicz, PWN, Warszawa-Poznań 1982) Kinetyka reakcji chemicznych zajmuje się badaniem szybkości przebiegu reakcji w zależności od różnych czynników (stężenia reagentów, temperatury, obecności katalizatorów). Zadaniem kinetyki jest ustalenie charakteru reakcji i matematyczne ujęcie zależności pomiędzy szybkością reakcji a czasem jej trwania, co pozwala na podanie szybkości reakcji w dowolnym czasie i przy dowolnym stężeniu substratu. Podstawą katalitycznej reakcji enzymatycznej jest odwracalne wzajemne oddziaływanie substratu (S) z enzymem (E), przy którym powstaje nietrwały kompleks (ES), który następnie rozpada się na enzym i produkt reakcji (P).
Schemat reakcji enzymatycznej można przedstawić równaniem:
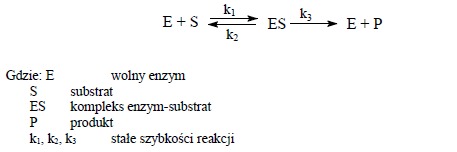
W równaniu tym zakłada się nieodwracalny mechanizm reakcji.
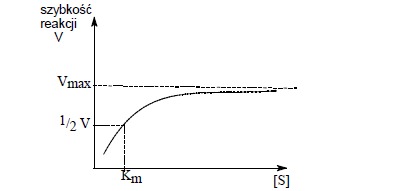
Przy małym stężeniu enzymu, lecz przy zmieniającym się w szerokich granicach początkowym stężeniu substratu, zmiany początkowej szybkości reakcji, wyrażone jako ilość substratu przetworzonego w jednostce czasu, można przedstawić w postaci krzywej:

Wpływ zwiększania stężenia substratu na szybkość reakcji enzymatycznej
Krzywa w pewnym punkcie zgina się i osiąga wartość maksymalną. Przy małym stężeniu substratu reakcja jest I rzędu i szybkość reakcji jest uzależniona od stężenia substratu [S].
Przy zwiększeniu stężenia substratu osiąga się maksymalną szybkość reakcji, która jest niezależna od stężenia substratu. Tłumaczy się to faktem, że przy zbyt małym stężeniu substratu niektóre cząsteczki enzymu nie są połączone z substratem w danym momencie i niecałkowite wysycenie enzymu jest przyczyną tego, że nie wykazuje on maksymalnej aktywności katalitycznej.
W miarę zwiększania stężenia substratu dochodzi do momentu, gdy wszystkie cząsteczki enzymu są z nim połączone i wówczas enzym pracuje z maksymalną szybkością. Dalsze zwiększanie stężenia substratu nie wpływa już na zwiększenie szybkości reakcji- reakcja przebiega ze stałą szybkością.
Dla reakcji (1) szybkość poszczególnych przemian jest następująca:

Wielkość Km zwana stałą Michaelisa jest wyrazem powinowactwa enzymu do substratu.
Enzymy o niskiej wartości Km działają znacznie sprawniej niż enzymy o wysokiej Km.
Szybkość tworzenia produktu v3 zależy od [ES], a więc im wartość [ES] jest większa, a tym samym stała Michaelisa mniejsza, tym szybsze powstawanie produktu.
Szybkość reakcji enzymatycznej można zapisać równaniem
v3 = v1-v2 (8)
Reakcja v2 przebiega bardzo wolno, więc można przyjąć, że szybkość procesu enzymatycznego v przebiega z szybkością v3:
v = v3 = k3[ES] (9)
Gdy substrat występuje w dużym nadmiarze, cały enzym jest nasycony substratem, [ES]=[Ec], szybkość reakcji osiąga swą wartość maksymalną:
Vmax = k3[Ec] (10)
Korzystając z odpowiednich przekształceń równań otrzymujemy zależność:
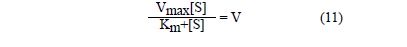
Graficzne przedstawienie równania Michaelisa-Menten
Jeżeli do równania (11) wstawimy v=vmax/2, to otrzymamy po przekształceniu:
Km = [S] (12)
Czyli stała Michaelisa odpowiada takiemu stężeniu substratu, przy którym szybkość reakcji wynosi połowę szybkości maksymalnej. Praktyczną metodą określania stałej Michaelisa i szybkości reakcji jest metoda graficzna.
Doprowadzając równanie (11) do postaci:
![]()
Otrzymujemy rónanie o postaci ogólnej y = ax + b, gdzie: b = 1/Vmax Równanie (13) nosi nazwę równania Lineweavera-Burka. Przekształcenie to ma na celu uzyskanie równania o ogólnej postaci linii prostej. W układzie współrzędnych wartości 1/v odkłada się na osi rzędnych, a wartości 1/[S] na osi odciętych. Równanie daje wówczas linię prostą, której nachylenie określa stosunek Km/Vmax. Przecina ona oś rzędnych w punkcie 1/Vmax, a oś odciętych w punkcie 1/Km. W ten sposób można określić maksymalną szybkość Vmax oraz stałą Michaelisa Km.
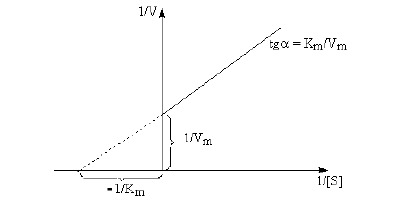
Graficzne przedstawienie równania Michaelisa-Menten według Lineweavera-Burka
Aktywność optyczna
Związki optycznie czynne posiadają zdolność skręcania płaszczyzny polaryzacji światła. Gdy światło spolaryzowane, wykazujące drgania w określonej płaszczyźnie przejdzie przez substancję optycznie czynną, wówczas jego drgania pojawiają się w innej płaszczyźnie. Kierunek i stopień skręcenia można mierzyć za pomocą polarymetru. Skręcenie w lewo oznaczamy znakiem (-), a skręcenie w prawo znakiem (+). Na przykład (+)-sacharoza jest prawoskrętna. Wielkość kąta skręcenia zależy od liczby cząsteczek znajdujących się na drodze wiązki światła podczas jej przechodzenia przez rurkę polarymetryczną, a zatem zależy od stężenia próbki i od długości rurki pomiarowej. Skręcalność właściwą [α]D danego związku definiujemy jako zaobserwowaną wartość skręcenia, gdy długość rurki pomiarowej wynosi 1 decymetr, stężenie próbki wynosi 1g/ml, a długość fali światła wynosi 589 nm.
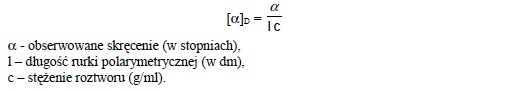
Skręcalność właściwa [α]D jest wielkością fizyczną charakterystyczną dla danego związku optycznie czynnego, podobnie jak temperatura topnienia, temperatura wrzenia, gęstość lub współczynnik załamania światła.
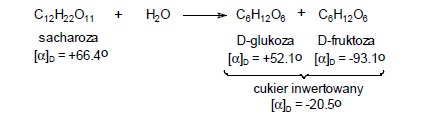
Hydroliza sacharozy
Sacharoza jest dwucukrem zbudowanym z cząsteczki D-glukopiranozy i D-fruktofuranozy. Wiązanie łączące glukozę z fruktozą ulega hydrolizie i to zarówno pod wpływem kwasów mineralnych, jak i enzymu zwanego inwertazą. Z tą reakcją związane jest zjawisko zwane inwersją sacharozy, polegające na zmianie znaku skręcalności roztworu sacharozy (+), gdyż w powstałej równomolowej mieszaninie cukrów fruktoza silniej skręca płaszczyznę polaryzacji w kierunku (-) niż glukoza (+) i cały roztwór przyjmuje skręcalność ujemną, czyli po reakcji znak skręcalności roztworu zmienia się na przeciwny (inwersja). Otrzymywany produkt, który jest mieszaniną D-glukozy i D-fruktozy nazywany jest cukrem inwertowanym.