
Autor: Anna Kurcek
Mitochondria są organellami komórkowymi przeprowadzającymi proces oddychania tlenowego. Przyjmuje się, że pochodzą one od bakterio-podobnych organizmów, które na drodze endosymbiozy zostały włączone do eukariotycznych komórek. Teorię tą potwierdza fakt, że mitochondria posiadają własny materiał genetyczny w postaci kilku (od 4 do 10) kopii kolistego nukleotydu o długość 16569 par zasad.
U ssaków, zawarta w nim informacja dziedziczona jest wyłącznie w linii żeńskiej. Dzieje się tak dlatego, że komórka jajowa posiada o wiele więcej cząsteczek mtDNA (ok. 100 000) niż plemniki (ok. 100), u których dodatkowo ulega on procesowi degradacji. Dzięki badaniom nad mitochondrialnym DNA pochodzącym od różnych grup etnicznych ustalono, że każdy współczesny Europejczyk jest potomkiem jednej z siedmiu kobiet żyjących w epoce kamiennej (od 10 do 50 tys. lat temu): Heleny, Veldy, Tary, Katriny, Ursuli, Yenii oraz Jasminy.
Sekwencję mtDNA poznano już w latach osiemdziesiątych. Wszystkie uzyskane dane dotyczące ludzkiego genomu mitochondrialnego zebrane są w bazie danych MITOMAP (http://www.mitomap.org/MITOMAP). Wiadomo, że zawiera on informację na temat 13 białek uczestniczących w procesie fosforylacji oksydacyjnej, 22 typów tRNA oraz 2 rodzajów rRNA (12S w małej i 16S w dużej podjednostce rybosomowej). Pozostałe białka zaangażowane w proces oddychania są kodowane przez DNA jądrowe, syntetyzowane w cytoplazmie i jako gotowe cząsteczki wnikają do mitochondriów.
Nici budujące podwójną helisę mtDNA określa się mianem ciężkiej H (zawiera informację na temat 12 białek i 14 tRNA) oraz lekkiej L (koduje 1 białko i 8 tRNA).
Kod genetyczny mitochondrialnego DNA różni się od uniwersalnego zapisu:
UGA – kodon STOP w mitochondriach oznacza tryptofan,
AUA – koduje metioninę zamiast izoleucyny,
AGA, AGG – kodujące argininę w mitochondriach oznaczają sygnał STOP.
Cechą charakterystyczną mtDNA jest również duże zagęszczenie kodowanych odcinków. Tzw. pętla D (ang. D-loop) jest jedynym fragmentem niezawierającym genów. Zachodzi w niej inicjacja transkrypcji. Odcinek ten zawiera dwa hiperzmienne obszary, które różnią się między sobą u poszczególnych osobników. Właściwość ta wykorzystywana jest w genetyce populacyjnej i medycynie sądowej.
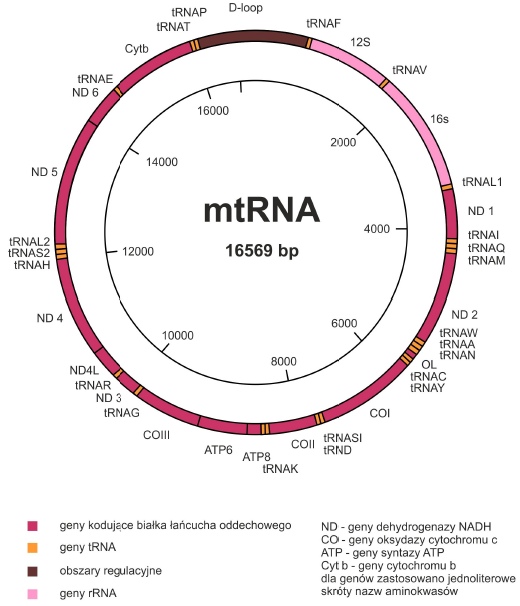
Rys. Ludzki mitochondrialny DNA
DNA mitochondrialny posiada mało sprawny system naprawczy i z tego powodu ewoluuje o wiele szybciej. Powstające zmiany mogą powodować zaburzenia funkcji fizjologicznych mitochondriów i prowadzić do występowania chorób, których objawy związane są przeważnie z tkankami o wysokim zapotrzebowaniu energetycznym – mięśniowej i nerwowej. Jednak ze względu na losową segregację mtDNA mogą dotyczyć właściwie wszystkich tkanek i narządów – szpiku kostnego, gruczołów i in.. Wielu chorobom towarzyszy zatem występowanie wielu różnych objawów klinicznych, które dodatkowo mogą się też różnić stopniem nasilenia u poszczególnych członków rodziny.
Przykładowe choroby:
• encefalopatia z kwasicą mleczanową i napadami udaropodobnymi (ang. mitochondria encephalopathy with lactic acidosis and stroke-like episodes, MELAS),
• zespół Lebera – dziedziczna neuropatia wzrokowa Lebera (ang. Leber’s hereditary optic neuropaty, LHON),
• zespół Kearnsa-Sayre’a (ang. Kearns-Sayre syndrome, KSS),
• porażenie mięśni odwodzących oka (ang. chronic progressive external ophtalmoplegia, CPEO),
• neuropatia obwodowa z ataksją i barwnikowym zwyrodnieniem siatkówki (ang. neurogenic muscle weaknes, atalia, retilis pigmentosa, NARP),
• padaczka miokloniczna z nieprawidłowymi czerwonymi włóknami mięśniowymi (ang. myoclonic epilepsy with ragged red fibres, MERFF).
Efekty mutacji pojawiają się najczęściej w dorosłym wieku, a ich leczenie jest jedynie objawowe.
Upośledzenie działania mitochondriów może być również wywołane zmianami w DNA jądrowym.
Literatura:
• Jerzy Bal „Biologia molekularna w medycynie. Elementy genetyki klinicznej”; Wydawnictwo Naukowe PWN; Warszawa 2001;
• Lubert Stryer „Biochemia”;Wydawnictwo Naukowe PWN; Warszawa 2003;
• Bryan Sykes „Siedem matek Europy”; Amber; 2002;
• Materiały z cytobiologii medycznej, www.histologia.cm-uj.krakow.pl/Cytobiologia/…/MITOCHON.DOC;
• Andreas M. Kogelnik, Marie T. Lott, Michael D. Brown, Shamkant B. Navathe and Douglas C. Wallace “MITOMAP: an update on the status of the human mitochondrial genome database”; Oxford University Press 1997;
• http://www.mitomap.org/MITOMAP.