
Choroby genetyczne stanowią bardzo różnorodną grupę schorzeń, wszystkie jednak powstają w wyniku mutacji DNA zaburzających prawidłowe funkcjonowanie organizmu. Tego typu zmiany mogą występować w pojedynczych genach lub też dotyczyć struktury i liczby całych chromosomów i być przekazywane z pokolenia na pokolenie.
Materiał genetyczny
Przez większość czasu DNA, czyli zlokalizowany w jądrze materiał genetyczny, występuje w postaci luźnej chromatyny. W trakcie podziałów przybiera ono jednak zbitą formę zwaną chromosomem w której każdy gen zostaje przyporządkowany ściśle określonemu miejscu, czyli tzw. locus. Ułatwia to znacznie rozdzielenie informacji między komórki potomne.
Każda prawidłowa ludzka komórka posiada zestaw 46 takich chromosomów, czyli 22 pary niezwiązanych z płcią autosomów oraz dwa chromosomy płciowe (XX u kobiet i XY u mężczyzn). Poszczególne pary mają charakterystyczną wielkość, kształt oraz układ prążków widocznych w wyniku różnych rodzajów barwienia. Odpowiednio ułożone tworzą one tak zwany kariotyp. Porównywanie go z materiałem genetycznym zdrowej osoby pozwala na wykrywanie i identyfikację niektórych nieprawidłowości leżących u podstaw chorób genetycznych. Wykonanie tego typu testów u płodu (tak zwana diagnostyka prenatalna) pozwala na wczesne rozpoznanie zaburzeń i umożliwia właściwe postępowanie w trakcie ciąży oraz przygotowuje rodziców na urodzenie chorego dziecka.
Rodzaje chorób genetycznych
Każdy rodzaj schorzenia genetycznego wynika z innego typu mutacji. Mogą się one różnić nawet między osobami dotkniętymi tym samym schorzeniem.
Ze względu na rodzaj powstałych zmian, choroby genetyczne dzieli się na trzy podstawowe kategorie:
• choroby powodowane przez defekty jednogenowe, lub inaczej monogenowe, które zachodzą w pojedynczych genach i wpływają na kodowaną przez nie informacje, prowadząc tym samym do wytworzenia zmienionego białka lub też zatrzymania jego produkcji;
• choroby wynikające z zaburzeń lub inaczej aberracji chromosomowych, zarówno liczbowych, jak i strukturalnych. Zmiana liczbowa dotycząca pojedynczego chromosomu nazywa się aneuploidią, natomiast obecność wielu kopii wszystkich chromosomów nosi nazwę poliploidii. Ta grupa schorzeń obejmuje także, powstające w wyniku złamań, delecje, insercje, duplikacje i rearanżację poszczególnych segmentów;
• zaburzenia wieloczynnikowe, uwarunkowane współdziałaniem wielu genów (poligenów) oraz warunków środowiskowych.
Defekty jednogenowe
Zmiany zachodzące w pojedynczych genach różnią się między sobą. Gdy dotyczą one pojedynczych nukleotydów, określa się je mianem mutacji punktowych. Do grupy tej należą m.in. mutacje sensu zmieniające kodon określonego aminokwasu i prowadzące do jego zamiany (substytucji) na inny monomer. Efekt tych mutacji zależy od tego, jaki charakter miał dany aminokwas (naładowany/ nienaładowany; hydrofilowy/hydrofobowy, duży/mały) oraz jak ważną rolę spełniał on w budowanym białku. Innym przykładem są tzw. mutacje nonsensowne, które prowadzą do zbyt wczesnego powstawania kodonu stop kończącego translację. Ich wpływ na kodowane białko jest tym poważniejszy, im bliżej znajdują się one końca 5’. To samo dotyczy tzw. mutacji ramki odczytu powstających w wyniku insercji lub delecji jednej lub kilku par zasad. Do mutacji punktowych zalicza się także zmiany miejsc splicingowych, które przebudowują sekwencje sygnalne splicingu znajdujące się na końcach 3’ i 5’ egzonów oraz w niekodujących częściach genomu, czyli tzw. intronach. Prowadzą one do powstawania nowych miejsc splicingowych oraz nieprawidłowego dojrzewania transkryptów. Na transkrypcję genu wpływają natomiast mutacje promotora. Występują one jednak dość rzadko.
Innym typem defektów jednogenowych są tzw. duże mutacje. Wyróżnia się wśród nich: delecje (polegające na utracie części normalnej sekwencji genu obejmującej od kilku zasad aż do całych sekwencji genu), insercje (gdy w normalnej sekwencji genu dochodzi do umieszczenia dodatkowego segmentu DNA pochodzącego z innego genomu) i rearanżacje (obejmujące obustronne przemieszczenia zachodzące pomiędzy segmentami sekwencji genu oraz innej części genomu).
Choroby wywoływane przez mutacje jednogenowe
Mutacje jednogenowe mogą powstawać de novo w komórkach somatycznych lub też rozrodczych, tzn. plemnikach i komórkach jajowych i być przekazywane z pokolenia na pokolenie zgodnie z prawami Mendla. Defekty zachodzące w pojedynczych genach są dziedziczone na kilka sposobów, który można określić przeprowadzając analizę rodowodu.
Mutacje autosomalne dominujące – dotyczą chromosomów niezwiązanych z płcią, a powodowana przez nie choroba ujawnia się nawet u osób posiadających tylko jeden nieprawidłowy wariant genetyczny, czyli u tzw. heterozygot. Według krzyżówek genetycznych, dzieci takich osób mają 50% szansy na odziedziczenie prawidłowej wersji genu. Choroby tego typu polegają najczęściej na uszkodzeniu białek strukturalnych, przenośnikowych lub receptorowych, a ich przebieg jest na ogół mniej ciężki niż w przypadku tzw. chorób recesywnych.
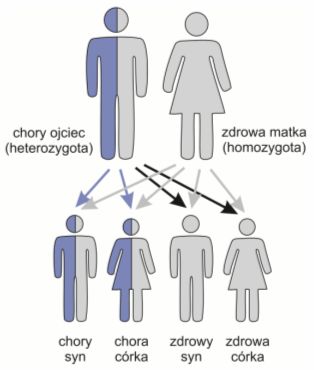
Rys. 1. Dziedziczenie chorób autosomalnych dominujących. Płeć dzieci nie ma znaczenia.
Należy przy tym pamiętać, że częstość ujawniania się cechy kodowanej przez dany gen, czyli tzw. penetracja może zależeć od różnych czynników, np. od warunków środowiska lub wieku chorego. Zjawisko to oraz zmienna ekspresja zmutowanego genu mogą zaburzyć obraz kliniczny w przypadku chorób autosomalnych dominujących.
Zestawienie 1. Przykłady chorób autosomalnych dominujących.
Rodzinna hipercholesterolemia – wysoki poziom LDL, czyli tzw. „złego cholesterolu”, występujący u całych rodzin przez wiele pokoleń, może prowadzić do wczesnego rozwinięcia się choroby wieńcowej i zawałów serca
choroba von Willebranda – jedna z najczęściej występujących skaz krwotocznych o łagodnym, umiarkowanym lub ciężkim przebiegu, spowodowana defektem lub niedoborem tzw. czynnika Willebranda, czyli jednego z białek umożliwiających krzepnięcie krwi
Autosomalnie dominująca wielotorbielowatość nerek (ADPKD) – schorzenie związane z mutacją genów PKD1 i PKD2 produkujących białka przez błonowe: policystynę 1 i 2, występujące w nabłonkach wielu narządów; do najistotniejszych objawów klinicznych należą torbiele na nerkach wywołujące nadciśnienie tętnicze oraz postępujące zmniejszanie się przesączania kłębuszkowego prowadzące do niewydolności nerek, występuje także torbielowatość wątroby oraz tętniaki mózgu wywołujące krwawienia podpajęczynówkowe
Pląsawica Huntingtona – schorzenie powodowane mutacją w genie HD (ang. huntingtin) i dotyczące ośrodkowego układu nerwowego; objawia się w postaci charakterystycznych i mimowolnych ruchów kończyn oraz twarzy, pojawiających się około 40-stego roku życia i stopniowo się wzmagających
Dystrofia miotyczna zwana również chorobą Steinerta lub Curshmanna-Steinerta – od innych dystrofii odróżnią ją charakterystyczny rozkład zaniku mięśni (głównie twarzy, szyi, przedramion, rąk, podudzia i stóp), występowanie miotonii, czyli opóźnionego rozkurczu mięśnia po uprzednim jego skurczu oraz występowanie zaburzeń niezwiązanych z mięśniami, np. zaćmy
Zespół Aperta – mutacja w genie FGF2 kodującym receptor 2 czynnika wzrostu fibroblastów, prowadzi do wad układu kostnego, obejmujących twarzoczaszkę – przedwczesne zarastanie szwów czaszkowych (kraniosynostoza) i ciasnotę wewnątrzczaszkową, a także rozszczep podniebienia i zrost palców dłoni i/lub stóp; u ok. 40% chorych występuje opóźnienie rozwoju psychoruchowego / niepełnosprawność intelektualna
Zespół Marfana – choroba tkanki łącznej o dużej zmienności fenotypowej, m.in. obejmuje objawy kostno- stawowe, wady układu krążenia i narządu wzroku
Achondroplazja – mutacje w genie FGFR3 objawiające się zaburzeniami rozwoju szkieletu (upośledzenie kostnienia śródchrzęstnego); osoby dotknięte chorobą charakteryzuje karłowatość, skrócenie długości kończyn w stosunku do długości tułowia, kyfoza odcinka lędźwiowego kręgosłupa, szpotawość kolan oraz charakterystyczny dysmorfizme twarzy
Retinoblastoma (siatkówczak) – wewnątrzgałkowy, złośliwy nowotwór jednego lub obu oczu występujący u dzieci, guz tworzy się w siatkówce; pierwszym objawem jest pojawienie się charakterystycznego odblasku w źrenicy (leukokoria) oraz zez.
Z chromosomami niezwiązanymi z płcią są związane również tzw. mutacje autosomalne recesywne. Wywoływane przez nie choroby występują jedynie w przypadku osób które odziedziczyły dwa zmutowane allele, czyli tzw. homozygot.
Heterozygoty, czyli osoby posiadające pojedynczy zmieniony allel, nie chorują, lecz mogą przekazywać uszkodzone geny swojemu potomstwu. Określa się je zatem mianem nosicieli.
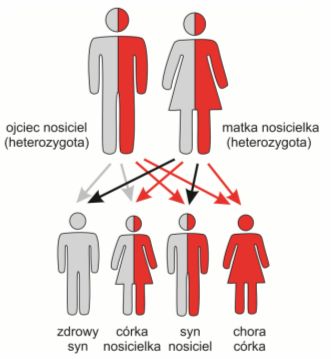
Rys. 2. Dziedziczenie chorób autosomalnych recesywnych, w rodzinie, w której oboje rodziców jest nosicielami uszkodzonego genu. Płeć dzieci nie ma znaczenia.
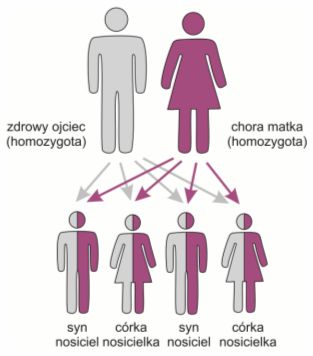
Rys. 3. Dziedziczenie zmutowanego allelu warunkującego chorobę autosomalną recesywną w rodzinie, w której jedno z rodziców jest chore, a drugie zdrowe. Płeć dzieci nie ma znaczenia.
Cechy dziedziczone w sposób recesywny występują o wiele rzadziej niż cechy dziedziczone w sposób dominujący i ujawniają się z jednakową częstotliwością u obu płci. Choroby tego typu dotyczą najczęściej defektów enzymatycznych.
W niektórych lokalnych populacjach częstość alleli recesywnych jest wyższa ze względu na pokrewieństwo jej członków. Jest to tzw. efekt założyciela.
Zestawienie 2. Choroby warunkowane przez mutacje autosomalne recesywne i występujące w grupach etnicznych.
Albinizm, lub inaczej bielactwo wrodzone – Indianie Hopi
schorzenie układu barwnikowego objawiające się brakiem pigmentu w skórze, tworach skórnych, włosach i tęczówce oka; spowodowane niedoborem enzymu tyrozynazy uczestniczącego w szlaku syntezy melaniny
Anemia sierpowata – Murzyni amerykańscy i afrykańscy, hindusi azjatyccy, mieszkańcy basenu Morza Śródziemnego (głównie Grecy); populacje Środkowego Wschodu
w wyniku obecności nieprawidłowej hemoglobiny – HbS, czerwone krwinki przybierają kształt sierpowaty i wykazują tendencję do zlepiania się; u chorych występuje zwiększona odporność na zarodźca malarii
β-talasemia – mieszkańcy basenu Morza Śródziemnego, populacje Bliskiego Wschodu, Hindusi, Chińczycy
powodowana przez ponad 100 różnych mutacji wywołujących zaburzenia w syntezie polipeptydów globiny i prowadzących do uszkodzeń czerwonych krwinek oraz anemii
Choroba Taya-Sachsa (TSD) – Żydzi aszkenazyjscy, Kanadyjczycy pochodzenia francuskiego
brak aktywnego enzymu beta heksozaminidazy A w lizosomach prowadzi do upośledzenia szlaków metabolicznych i nagromadzenia się złogów niemetabolizowanych substratów zaburzających funkcjonowanie komórki i całego układu nerwowego; choroba jest śmiertelna, a pierwsze objawy kliniczne występują w wieku od 3 do 9 miesięcy
Choroba Gauchera – Żydzi aszkenazyjscy
zaburzeniem funkcji lizosomów związane z mutacją genu GBA zlokalizowanego na chromosomie 1; wyróżnia się trzy typy choroby różniące się od siebie przebiegiem oraz intensywnością występowania objawów klinicznych, takich jak np. powiększona wątroba i śledziona, osteoporoza i niedokrwistość
Zespół Blooma – Żydzi aszkenazyjscy
spowodowana mutacją genu BLM występującego na 15 chromosomie, charakteryzuje się rozszerzeniem drobnych naczyń krwionośnych, wrażliwością na światło, niskorosłością, niedoborem odporności oraz zwiększoną podatnością na nowotwory
Ciężki złożony defekt odporności (SCID) – Indianie Apacze
zablokowanie wzrostu i namnażania się limfocytów T i B upośledza układ odpornościowy i objawia się już w dzieciństwie podatnością na choroby, m.in. biegunki, pleśniawki, zakażenia skóry i ucha oraz zapalenie płuc; objawom towarzyszy niski wzrost i masa ciała
Fenyloketonuria – Europejczycy
choroba metaboliczna, której przyczyną są mutacje w genie kodującym enzym uczestniczący metabolizmie fenyloalaniny (hydroksylazę fenyloalaninową, PAH); nagromadzenie się tego aminokwasu w organizmie ma działanie toksyczne; wczesne rozpoznanie może zapobiec rozwinięciu się powikłań, np. uszkodzeniu ośrodkowego układu nerwowego.
Więcej na temat fenyloketornurii dowiecie się pod adresem www.vitapku.pl
Mukowiscydoza – Europejczycy
u chorych występuje nieprawidłowy skład wydzieliny gruczołów zewnątrzwydzielniczych; gęsta i kleista wydzielina zalega w gruczołach i może powodować niedrożność przewodów wyprowadzających, w efekcie czego może dochodzić do włóknienia niektórych narządów (wątroby, trzustki, oskrzeli); jednym z pierwszych i najcięższych objawów mukowiscydozy jest tzw. niedrożność smółkowa będąca wynikiem niedoboru enzymów trzustkowych – smółka, czyli zawarość jelit noworodka zatyka światło jelita; występują również stany zapalne dróg oddechowych
Wrodzony zespół nerczycowy typu fińskiego – Finowie
wywołany przez mutacje genu związanego z produkcja nefryny należącej do rodziny komórkowych cząsteczek adhezyjnych; już w życiu płodowym pojawia się białkomocz, czyli nadmierna utrata białka z moczem
Wrodzony przerost nadnerczy, lub inaczej zespół nadnerczowo-płciowy – Eskimosi
niedobór enzymów zaangażowanych w proces produkcji hormonów nadnerczy (głównie 21-hydroksylazy), prowadzący do niedoczynności nadnerczy oraz nadprodukcji męskich hormonów płciowych
Innymi przykładami takich chorób mogą być:
• galaktozemia – mutacja w genie GALT warunkuje brak enzymu uczestniczącego w metabolizmie galaktozy, prowadząca do nagromadzenia się tego cukru w organizmie. Do objawów należą opóźniony rozwój, powiększona wątroba, zaćma oraz opóźnienie umysłowe;
• homocystynuria – mutacja genu CBS kodującego enzym β-cystationionową, zaburzająca metabolizm siarki. Nieleczona może prowadzić do krótkowzroczności, nieprawidłowości szkieletowych, opóźnienia umysłowego, a także zdarzeń zakrzepowo-zatorowych;
• rdzeniowy zanik mięśni (SMA) – mutacje genu SMN1 prowadzące do zaniku motoneuronów rdzenia kręgowego, prowadzące do osłabienia i atrofii tkanki mięśniowej.
Nie wszystkie choroby dotyczą jednak autosomów. Cześć defektów genetycznych może być sprzężona z chromosomem płciowym X. Tego typu choroby mogą być zarówno recesywne, jak i dominujące.
Choroby recesywne najczęściej dotyczą mężczyźni, ponieważ posiadają oni tylko jeden chromosom X, to znaczy są tzw. hemizygotami i w związku z tym do ujawnienia się choroby potrzeba u nich tylko jednego allelu. Objawy tego typu schorzeń rzadko pojawiają się u kobiet (XX), które najczęściej pozostają nosicielkami.
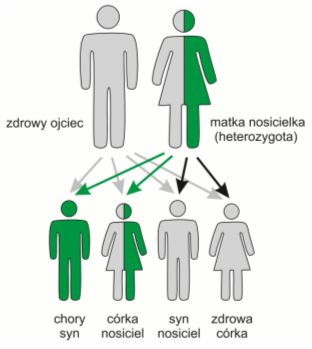
Rys. 4. Dziedziczenie mutacji recesywnych związanych z chromosomem X. Gdy matka jest nosicielką statystycznie rzecz biorąc połowa jej męskiego potomstwa jest chora, a połowa dzieci płci żeńskiej zostaje nosicielkami. Choroba może być również przekazywana córkom przez chorych mężczyzn.
Przykładem tego typu chorób może być dystrofia mięśniowa Duchenne’a (DMD), w wyniku której już w dzieciństwie dochodzi do zaniku mięśni. Około 10 roku życia chory przestaje chodzić, natomiast śmierć występuje około 20 roku życia w wyniku niewydolności sercowej i oddechowej. Choroba ta może wystąpić również u kobiet w wyniku nietypowej inaktywacji X (gdy w większości komórek mięśniowych inaktywowany jest prawidłowy X. Objawy te są jednak łagodniejsze niż u mężczyzn), nowej mutacji na drugim chromosomie X nosicielki i zespołu Turnera (objawy są wtedy równie ciężkie co w przypadku mężczyzn), a także translokacji chromosomu X z mutacją na autosom, w wyniku której dochodzi do inaktywacji prawidłowego chromosomu X.
Zestawienie 3. Przykłady chorób warunkowanych mutacją recesywną sprzężoną z chromosomem X.
Daltonizm – może być wynikiem uszkodzenia siatkówki oka lub też uszkodzenia dróg wzrokowych i polega na nierozróżnianiu barwy czerwonej (tzw. Protanopia), zielonej (tzw. Duoteranopia) oraz żółto-niebieskiej (tzw. Tritanopia)
Dystrofia mięśniowa Beckera (BMD) – mutacja w genie dystrofiny powodująca nieodwracalny zanik mięśni; choroba ma charakter postępujący, a pierwsze jej objawy występują w wieku 5–25 lat i obejmują mięśnie szkieletowe, potem także mięsień sercowy, co prowadzi do kardiomiopatii; śmierć następuje najczęściej w wyniku niewydolności oddechowej lub układu krążenie w wieku około 45 lat
Hemofilia – skaza osoczowa polegająca na niedoborze czynnika krzepnięcia VIII (typ A) lub IX (typ B) i powodująca wydłużone krwawienia; objawy kliniczne obu typów choroby różni się od siebie intensywnością
Rybia łuska sprzężona z chromosomem X – nadmierne nagromadzenie się i wytwarzanie zrogowaciałych komórek naskórka, mogą się także pojawiać objawy rogowacenia mieszkowego
Zespół kruchego (łamliwego) chromosomu X (zespół fra X) – obecność zaburzenia w liczbie sekwencji powtarzalnych (trójki nukleotydów – CGG) eksonu 1 genu FMR1, jest to mutacja dynamiczna, ponieważ liczba powtórzeń wzrasta w każdym pokoleniu; jej pierwszym objawem klinicznym jest upośledzenie umysłowe, a u mężczyzn także charakterystyczny wygląd (podłużna twarz, guzy czołowe, duża żuchwa, odstające małżowiny uszne)
Choroby związane z <strong.mutacją dominującą i sprzężoną z chromosomem X występują zarówno u mężczyzn, jak i u kobiet. U wszystkich mężczyzn przebiegają one równie ciężko. U kobiet efekt ten zależy od pionizacji, czyli od losowego wyłącznie jednego z heterosomów.
Wyróżnia się wśród nich:
• krzywica hipofosfatemiczna witamino-D-oporna (hiperfosfatemia rodzinna) – nieprawidłowa mineralizacja tkanki kostnej uwarunkowana genetycznym defektem wchłaniania fosforanów w nerkach; do charakterystycznych objawów należą zmiany krzywicze w kościach oraz niewrażliwość na leczenie witaminą D;
• zespół Retta – zaburzenie rozwoju często mylnie diagnozowane jako autyzm, choroba dotyczy jedynie dziewczynek (u mężczyzn cecha jest letalna, tzn. prowadzi do poronień), cechuje się zaburzeniami ruchów dłoni i ciała, apraksją, czyli upośledzeniem precyzyjnych, celowych ruchów, brakiem komunikacji słownej, zaburzenia kontaktu wzorkowego oraz spowolnieniem przyrostu obwodu głowy;
• zespół Blocha- Sulzbergera (nietrzymanie barwnika) – stwierdza się jedynie u heterozygotycznych dziewczynek, u mężczyzn zmiany są rozległe i prowadzą do poronień. W okresie noworodkowym a czasami już w życiu płodowym pojawiają się zmiany skórne, które wraz z wiekiem ulegają charakterystycznej ewolucji. Zaburzenia występują też w obrębie przydatków skóry, narządzie wzroku, układzie kostnym i w ośrodkowym układzie nerwowym.

Rys. 5. Dziedziczenie chorób warunkowanych mutacją dominującą sprzężoną z chromosomem X w rodzinie, w której ojciec jest chory, a matka zdrowa.
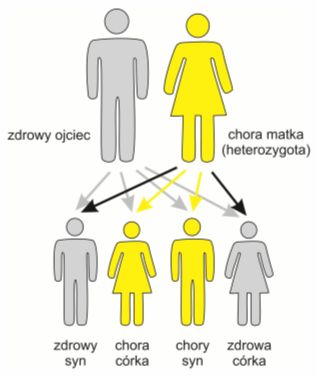
Rys. 6. Dziedziczenie chorób warunkowanych mutacją dominującą sprzężoną z chromosomem X w rodzinie, w której matka jest chora, a ojciec zdrowy.
Dziedziczenie mitochondrialne
Mitochondria są to centra energetyczne komórki znajdujące się w dużej ilości w ośrodkowym układzie nerwowym, mięśniach, nerkach i gruczołach wydzielania wewnętrznego, czyli tkankach wykazujących duże zapotrzebowanie energetyczne. Posiadają one własny materiał genetyczny, czyli tzw. mitochondrialne DNA (mtDNA). Ma ono o wiele słabszy system naprawczy niż DNA jądrowe i najczęściej zachodzą w nim mutacje punktowe i zmiany długości sekwencji nukleotydowej. Choroby mitochondrialne mogą również spowodowane mutacjami w jądrowym DNA kodującym białka specyficzne dla mitochondriów oraz związane z regulacją ich funkcjonowania.
Do tej grupy schorzeń należą głównie miopatie, kardiomiopatie i zaburzenia neurologiczne. Ich efekt fenotypowy wykazuje dużą zmienność i zależy od liczby cząsteczek DNA z mutacją. Choroby tego typu dziedziczone są tylko po matce ale przez dzieci obu płci.
Zestawienie 4. Przykłady chorób dziedziczonych mitochondrialnie.
Zespół przewlekłej postępującej zewnętrznej oftalmoplegii, (CPEO) – powodowany delecją w modna; do jego objawów należy przewlekła zewnętrzną oftalmoplegia (porażenie mięśni gałki ocznej), obustronne opadanie powiek oraz osłabienie mięśni okołoruchowych
Zespół Kearnsa-Sayre’a (KSS) – przyczyną jest delecja w mtDNA; choroba nie jest dziedziczona; objawy występują przed 20 rokiem życia i obejmują postępującą oftalmoplegię zewnętrzną, zwyrodnienie barwnikowe siatkówki, ataksję, podwyższony poziom białka w płynie mózgowo-rdzeniowym, zaburzenia przewodzenia w mięśniu sercowym; nie stwierdza się przy tym osłabienia kończyn
Zespół mitochondrialnej encefalomiopatii dotyczącej układu nerwowego, żołądka i jelit (MNGIE) – spowodowany mutacją w genie znajdującym się na chromosomie 22 i kodującym fosforylazę tymidyny oraz delecjami w mtDNA; objawy występują między 20 a 50 rokiem życia i zalicza się do nich opadanie powiek, oftalmoplegię, zaburzenia motoryki przewodu pokarmowego, miopatię mięśni szkieletowych i obwodową neuropatię
Zespół szpikowo-trzustkowy Pearsona – wywołuje go pojedyncza delecja w mtDNA; objawy to ciężka niedokrwistość makrocytowa w pierwszych tygodniach życia, podwyższony poziom hemoglobiny, zmienna neutropenia i trombocytopenia, zaburzenia pracy trzustki, cukrzyca, zaburzenia funkcji wątroby, zanik kosmków jelitowych, zaburzenia krzepnięcia krwi
Padaczka miokloniczna z występowaniem „włókien szmatowatych” w mięśniach, zespół MERRF – spowodowana głównie mutacją punktową nukleotydu 8344 G-A w genie kodującym tRNA lizyny w mtDNA; objawy choroby mogą występować w wieku późnodziecięcym lub dojrzałym i obejmują padaczkę, ataksję i miopatię z występowaniem włókien szmatowatych w badaniu biopsji mięśni oraz podwyższony poziom kwasu mlekowego, postępujące otępienie, zanik nerwu wzrokowego i utratę słuchu
Zespół MELAS (miopatia mitochondrialna, encefalopatia, kwasica mleczanowa, występowanie incydentów podobnych do udarów) – przyczyną choroby jest głównie mutacja punktowa nukleotydu 3243 w obrębie genu tRNA leucyny w mtDNA; objawy pojawiają się w dzieciństwie i obejmują miopatię z encefalopatią, incydenty udaropodobne, kwasicę mleczanową, cofanie się umiejętności psychoruchowych, drgawki, głuchotę, zwyrodnienie barwnikowe siatkówki, cukrzycę i niski wzrost
Dziedziczna neuropatia nerwu wzrokowego Lebera, LHON – zanik nerwów wzrokowych spowodowany różnymi mutacjami modna; chorują zwykle mężczyźni (u kobiet objawy pojawiają się później i są łagodniejsze), choroba ujawnia się przeważnie między 10 a 20 rokiem życia i dotyczy obu oczu jednocześnie, utrata wzroku następuje w ciągu 8 tygodni, chorobie mogą towarzyszyć zmiany podobne do objawów stwardnienia rozsianego, neuropatia obwodowa, ataksja oraz encefalopatia
,br> Neurogenna miopatia z ataksją i zwyrodnieniem barwnikowym siatkówki, zespół NARP – wywołana punktową mutacją w nukleotydzie 8993 w mtDNA genu kodującego syntazę ATP, do objawów zalicza się zwyrodnienie barwnikowe siatkówki, demencję, drgawki, ataksję, osłabienie mięśni proksymalnych, neuropatię czuciową, a niekiedy także opóźnienie rozwoju lub dysfunkcję pnia mózgu, zaburzenia czynności wątroby i przewlekłą kwasicę
Zatrucie wywołane podaniem chloramfenikolu – punktowe mutacje w genie kodującym podjednostkę 16S rRNA powodują u 1 na około 19.000 osób w zdrowej populacji objawy toksyczne po podaniu leczniczej dawki chloramfenikolu
Aberracje chromosomowe
Zaburzenia struktury chromosomów powstają zazwyczaj de novo w rodzicielskich komórkach rozrodczych. Mogą być one wynikiem pęknięcia chromosomu i powstania rekombinacji o nieprawidłowej konfiguracji. Dość często spotyka się tzw. translokacje wzajemne, czyli wymianę segmentów między niehomologicznymi chromosomami. Jeżeli są one zrównoważone, nie prowadzą powstawania zaburzeń, ale mogą powodować u potomstwa groźne translokacje niezrównoważone (połączone ze zmianą ogólnego składu genowego).
Inny typem zmian jest translokacja robertsonowska, czyli fuzja długich ramion chromosomów akrocentrycznych (13, 14, 15, 21 i 22) z jednoczesną utratą ramion krótkich. Są one pozbawione istotnego materiału genetycznego i dlatego ich strata nie jest groźna. Może ona jednak prowadzić do powstawania u potomstwa trisomii, czyli obecności trzech kopii danego chromosomu, np. translokacja robertsonowska chromosomów 14 i 21 i jest odpowiedzialna za około 5% przypadków zespołu Downa.
Do innych typów niezrównoważonych zaburzeń chromosomowych należą delecje, czyli utraty odcinków chromosomów. Niektóre z nich trudno jest zauważyć podczas rutynowych badań kariotypu. Są one jednak przyczyną wielu różnych zespołów mikrodelecji lub inaczej delecji genów sąsiadujących, np. zespół mikrodelecji 22q, stanowiący częstą przyczynę wrodzonych wad serca.
Duplikacje odpowiednich odcinków chromosomów powodują natomiast powstanie częściowej trisomii. Tak zwane chromosomy markerowe (ang. extra structurally abnormal chromosomes, ESAC), są to dodatkowe, małe i niepodobne do innych chromosomów struktury. Wyróżnia się także chromosomy pierścieniowe, inwersje paracentryczne lub pericentryczne, a także izochromosomy, czyli chromosomy zawierające jedynie dwie kopie pojedynczego ramienia.
Najczęściej spotykanym u ludzi zaburzeniem chromosomowym o znaczeniu klinicznym jest aneuploidia, czyli nieprawidłowa liczba chromosomów, np. trisomia, której prawdopodobieństwo wystąpienia wzrasta wraz z wiekiem matki.
Zestawienie 5. Przykłady chorób wywołanych trisomią.
Zespół nadkobiety – powstaje, gdy jedna z gamet zawiera dwa chromosomy X; chora kobieta ma bardzo silnie zaznaczone cechy płciowe, niski wskaźnik inteligencji i obniżoną płodność
Zespół Klinefeltera – dotyczy mężczyzn posiadających dodatkowy chromosom X (konfiguracja 44+ XXY), który wywiera wpływ feminizujący, objawiający się w brzmieniu głosu, owłosieniu, układzie tkanki tłuszczowej, rozwoju sutków oraz powoduje niedorozwój jąder i prącia; zmianom towarzyszy bezpłodność; poziom inteligencji normalny, możliwe są jednak zaburzenia seksualne oraz agresywność
Zespół nadmężczyzny – powstaje, gdy plemnik zawiera dwa chromosomy Y; chorzy odznaczają się wysokim wzrostem i agresywnością, pozostają jednak płodni, a ich potomstwo jest zdrowe
Zespół Downa – trisomia chromosomów 21 pary (45 +XX lub 45+ XY), występująca raz na 600-800 urodzeń; chore osoby rysami twarzy przypominają rasę mongolską (owalne, skośne ku dołowi szpary powiekowe, duży wystający język, małe uszy, opadające kąciki ust); obserwuje się także zmiany w proporcji budowy ciała oraz zaburzenia umysłowe, jak również wady serca, opóźnione dojrzewanie płciowe i zwiększone ryzyko wystąpienia białaczki; nowonarodzone dzieci wykazują zazwyczaj wiotkość mięśni, są jednak pogodne i przywiązane do rodziców
Zespół Edwardsa – trisomia chromosomu 18, występuje raz na 10000 noworodków, trzy razy częściej u dziewczynek; objawia się napięciem mięśniowym, przykurczem kończyn, suszkowatymi, czyli płaskimi stopy (kształt suszki używanej dawniej do osuszania atramentu) oraz zaciskanymi piąstkami w przypadku niemowląt, w niektórych przypadkach obserwuje się również wady serca oraz ośrodkowego układu nerwowego
Nieco rzadziej występują tzw. monosomie, gdyż są o wiele częściej letalne, to znaczy prowadzą do śmierci zarodka. Przykładem może być zespół Turnera, czyli monosomia kobiet spowodowana nieobecnością jednego z chromosomów X (44+X). Choroba może dotyczyć wszystkich komórek organizmu lub niektórych z nich (mozaicyzm). Powoduje bezpłodność, niedorozwój jajników, pochwy i macicy oraz brak piersi i owłosienia łonowego oraz pachowego. Zmianom towarzyszy zazwyczaj upośledzenie umysłowe, a także inne nieprawidłowości, takie jak niski wzrost, nieprawidłowe ukształtowanie małżowiny usznej, wady oczu, opadające powieki, zwężenie tętnicy głównej, niska linia włosów na karku, szeroko rozstawione brodawki sutkowe.
Choroby dziedziczone poligenowo
W przypadku chorób dziedziczonych poligenowo ważną rolę odgrywają nie tylko czynniki genetyczne ale również warunki środowiskowe i w związku z tym wczesne wykrycie tego typu nieprawidłowości umożliwia właściwą profilaktykę, np. dietę lub ćwiczenia. Stopień ich wpływu na kształtowanie się danej cechy można ocenić dzięki badaniom na bliźniętach jedno i dwujajowych.
Ryzyko wystąpienia tego typu chorób zależy m.in od stopnia zaawansowania choroby przodka, liczby członków rodziny dotkniętych chorobą i stopnia łączącego ich pokrewieństwa, a także liczby genów warunkujących daną cechę.
Przykłady chorób:
• rozszczep wargi i podniebienia
• rozszczep kręgosłupa
• zwężenie odźwiernika
• choroba niedokrwienna serca
• nadciśnienie tętnicze
• cukrzyca
• wrodzona wada serca
• padaczka
• schizofrenia
• psychoza maniakalno-depresyjna
• wrodzone zwichnięcie stawu biodrowego
• choroba wrzodowa
• choroby alergiczne
• reumatoidalne zapalenie stawów
Autor: Anna Kurcek
Literatura:
1. Sadakiersa-Chudy A., Dąbrowska G., Goc a., 2004. Genetyka ogólna. Skrypt do ćwiczeń dla studentów biologii. Uniwersytet Mikołaja Kopernika Toruń.
2. Strona Krajowego centrum Diagnostyki i Leczenia Hipercholesterolemii Rodzinnej. http://www.hipercholesterolemia.com.pl/
3. Czym jest choroba vonWillebranda? http://www.idn.org.pl/hemofilia/chorobavWillebranda.pdf
4. Różański J., Ciechanowski K., 2010. Autosomalnie dominująca wielotorbielowatość nerek. Fakty i mity. Forum Nefrologiczne 3 (3): 143–149. http://czasopisma.viamedica.pl/fn/article/download/10372/8842
5. Dystrofia miotyczna. Broszura wydana przez Towarzystwo Zwalczania Chorób Mięśni. www.ptchnm.slask.pl/wt/doc/Dystrofia_miotoniczna.doc
6. Centrum Genetyki Medycznej Genesis. http://www.genesis.pl/
7. http://retinoblastoma.com/retinoblastoma/
8. http://www.zdronet.pl/
9. Strona Stowarzyszenia na Rzecz Dzieci z Zaburzeniami Genetycznymi. http://www.gen.org.pl/
10. Maruniak-Chudek I., Niemir Z.I., Świetliński J., 2004. Podłoże genetyczne zespołów nerczycowych u dzieci. Postepy Hig Med Dosw. 58: 405-415. http://www.phmd.pl/fulltxt.php?ICID=448931
11. Stowarzyszenie pacjentów z wrodzonym przerostem nadnerczy. http://www.wpn.org.pl/
12. Strona Polskiego Towarzystwa Chorób Nerwowo-Mięśniowych (PTChNM) http://ptchnm.org.pl/
13. Strona Ogólnopolskiego Stowarzyszenia Pomocy Osobom z Zespołem Retta. http://www.rettsyndrom.gd.pl/
14. Urbaniak N., Czyżyk E., Mazur A., Korczowski B., 2012. Zespół Blocha-Sulzbergera. Przegląd Medyczny Uniwersytetu Rzeszowskiego i Narodowego Instytutu Leków w Warszawie Rzeszów. http://www.pmurz.rzeszow.pl/PDF/2012/1/16a_.pdf
15. http://www.ochoroba.pl
16. http://www.genetyka.xzq.pl/
17. Norton M. E., Genetyka i diagnostyka prenatalna.
18. http://www.elsevier.pl/layout_test/book_file/58/ultrasonografiacallen-rozdzial2.pdf