PRZEDRUK, oryginał dostępny pod adresem www
Tytuł oryginalny: Materiały dodatkowe – związki karbonylowe
Autor: dr Marek Żylewski
Uniwersytet Jagielloński (www)
Wydział Farmaceutyczny Collegium Medicum (www)
Katedra Chemii Organicznej (www)
Kierownik: Prof. UJ, dr hab. Marek Cegła
Adres:
ul. Medyczna 9
30-688 Kraków
Kontakt: tel. 012 620 55 00
1. Polaryzacja wiązania C=O
Charakterystyczną cechą związków posiadających grupę karbonylową (dotyczy to nie tylko aldehydów czy ketonów, ale też np. dowolnych pochodnych kwasów karboksylowych) jest silna polaryzacja wiązania C=O, dla którego, dzięki zjawisku mezomerii, zdefiniować można drugą strukturę z rozdzielonym ładunkiem:
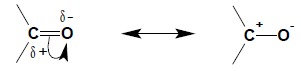
Polarna budowa grupy karbonylowej z atomem węgla obdarzonym cząstkowym ładunkiem dodatnim (należy pamiętać, iż struktury mezomeryczne obrazują krańcowe możliwości przesunięcia par elektronowych, natomiast rzeczywista struktura cząsteczki jest hybrydą wszystkich możliwych struktur granicznych) determinuje reaktywność chemiczną tej grupy i kierunek reakcji związków karbonylowych z odczynnikami nukleofilowymi, których przykłady podano poniżej.
2. Addycja wody – otrzymywanie wodzianów
Charakterystyczne dla chemii związków karbonylowych są reakcje addycji do wiązania podwójnego grupy karbonylowej różnych reagentów. Najprostszym przykładem jest reakcja addycji cząsteczki wody, zachodząca jako proces równowagowy w wodnych roztworach aldehydów czy ketonów:
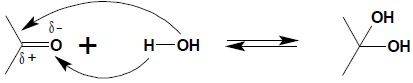
Otrzymanych wodzianów wyodrębnić w stanie czystym nie można poza nielicznymi przypadkami, w których obecność dwóch grup hydroksylowych przy jednym atomie węgla jest stabilizowana poprzez silnie elektroujemny podstawnik w bezpośrednim sąsiedztwie grupy karbonylowej. Należy do nich wodzian chloralu – związek znajdujący zastosowanie w weterynarii jako środek usypiający:
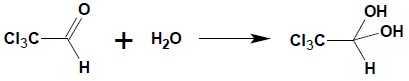
3. Reakcja związków karbonylowych z alkoholami.
Związki karbonylowe reagują z alkoholami podobnie jak z wodą, przyłączając cząsteczkę alkoholu do wiązania podwójnego zgodnie z rozkładem ładunku w grupie karbonylowej:
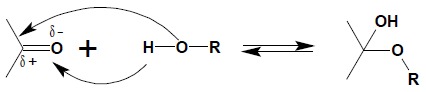
Powstały produkt nosi nazwę hemiacetal (jeśli wyjściowym związkiem karbonylowym był aldehyd) lub hemiketal – jeśli powstał z ketonu. Reakcja powyższa jest procesem odwracalnym, katalizowanym zarówno przez kwasy, jak i zasady. W środowisku kwaśnym możliwa jest dalsza reakcja hemiacetali czy hemiketali z alkoholem prowadząca do powstania acetalu lub ketalu:
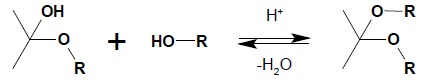
Reakcja ta jest reakcją substytucji nukleofilowej przy karbonylowym atomie węgla – w miejsce grupy OH hemiacetalu czy hemiketalu wchodzi reszta OR alkoholu. Proces ten ma bardzo duże znaczenie w chemii cukrów, gdzie jest odpowiedzialny za tworzenie się pierścieniowych form cukrów i powstawanie wiązań pomiędzy jednostkami cukrowymi w oligo i polisacharydach. W syntezie organicznej duże znaczenie ma tworzenie się pierścieniowych acetali czy ketali, co jest wykorzystywane jako ochrona grupy karbonylowej w przypadku, kiedy trzeba wykonać reakcję w innym miejscu cząsteczki bez naruszenia grupy karbonylowej związku. Pierścieniowe acetale bądź ketale powstają w wyniku reakcji związku karbonylowego i odpowiedniego diolu (najłatwiej powstają pierścienie 5-cio i 6-cio członowe) w środowisku kwaśnym:
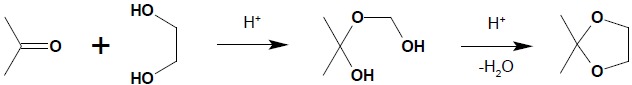
Powstałe pierścieniowe acetale lub ketale łatwo ulegają hydrolizie w środowisku kwaśnym, natomiast są stabilne w środowisku zasadowym, zatem ochrona grupy karbonylowej poprzez przeprowadzenie jej w pierścieniowy acetal lub ketal jest skuteczna w reakcjach substytucji nukleofilowej czy utlenienia przebiegających w środowisku zasadowym, czy obojętnym.
4. Reakcja związków karbonylowych z HCN
Aldehydy i ketony w reagują z cyjanowodorem analogicznie jak z wodą, dając cyjanohydryny:
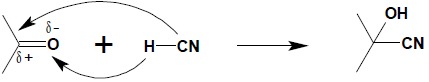
Ze względu na łatwość przeprowadzenia grupy cyjanowej w grupę karboksylową reakcja ta ma duże znaczenie w syntezie organicznej jako etap pośredni otrzymywania α-hydroksykwasów.
5. Azotowe pochodne aldehydów i ketonów – reakcja ze związkami posiadającymi grupę NH2
Kolejnym przykładem addycji nukleofilowej do grupy karbonylowej aldehydów i ketonów jest reakcja tych związków z aminami I-rzędowymi:
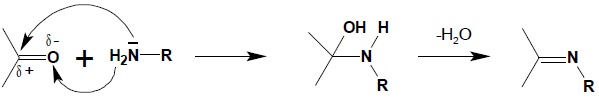
Powstały w pierwszym etapie reakcji aminoalkohol jest związkiem nietrwałym, ulegającym natychmiastowej dehydratacji z utworzeniem iminy. Oprócz amin I-rzędowych w reakcji tej często stosuje się różne pochodne hydrazyny (H2N-NH2) otrzymując szereg połączeń azotowych aldehydów i ketonów:
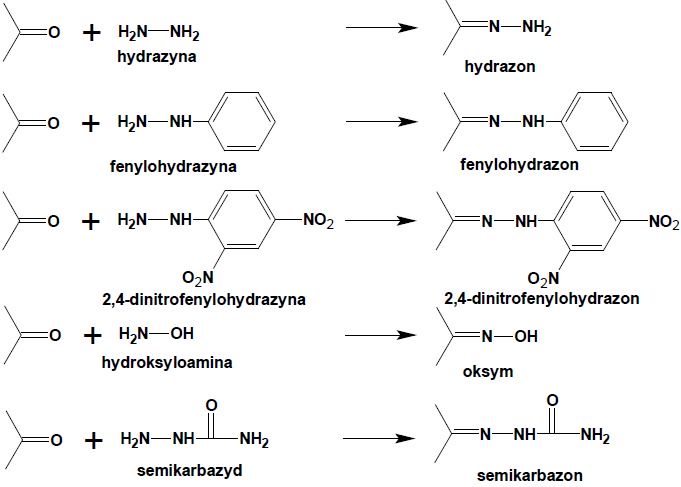
Hydrazony i oksymy znajdują zastosowanie w syntezie organicznej, natomiast fenylohydrazony, 2,4-dinitrofenylohydrazony i semikarbazony związków karbonylowych służą jako tzw. stałe pochodne w analizie aldehydów i ketonów
6. Reakcja związków karbonylowych z aminami II-rzędowymi – otrzymywanie enamin Aminy drugorzędowe również mogą być substratami w reakcji addycji do grupy karbonylowej, jednakże ze względu na obecność jedynie jednego atomu wodoru w grupie NH, eliminacja cząsteczki wody nie jest możliwa w sposób, w jaki zachodzi ona w reakcji związków karbonylowych z aminami I-rzędowymi. Aby mógł powstać trwały produkt reakcji pomiędzy aldehydem czy ketonem a aminą II-rzędową (układy, w których przy jednym atomie węgla obecne są dwa silne elektroujemne podstawniki, takie jak np. grupy OH, NR2 są układami nietrwałymi) konieczna jest eliminacja cząsteczki wody z udziałem protonu związanego z α atomem węgla związku karbonylowego:
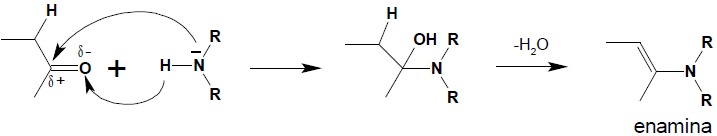
W wyniku reakcji powstaje układ, w którym trzeciorzędowa grupa aminowa jest związana z atomem węgla stojącym przy wiązaniu podwójnym – enamina. Warunkiem koniecznym, aby możliwa była reakcja związku karbonylowego z aminą II-rzędową jest zatem obecność atomu wodoru w pozycji α związku karbonylowego.
7. Addycja NaHSO3
Kolejnym przykładem reakcji addycji do spolaryzowanego wiązania C=O grupy karbonylowej jest reakcja z NaHSO3:
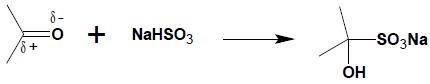
W wyniku otrzymuje się sole sodowe kwasów α-hydroksysulfonowych, zwane zwyczajowo połączeniami bisulfitowymi. Związki te najczęściej są krystalicznymi ciałami stałymi, często trudno rozpuszczalnymi w wodzie, dzięki czemu mogą służyć do wydzielania aldehydów i ketonów z mieszanin. W syntezie organicznej służą również jako mniej reaktywne pochodne związków karbonylowych (zastosowanie ich pozwala np. na uniknięcie reakcji kondensacji benzoinowej, zachodzącej jako reakcja uboczna, podczas przyłączenia HCN do aldehydów aromatycznych). Zaznaczyć należy, iż ketony aromatyczno-alifatyczne oraz ketony aromatyczne nie reagują z NaHSO3.
8. Reakcja aldehydów i ketonów ze związkami Grignarda
Bardzo ważną z punktu widzenia syntezy organicznej reakcją addycji nukleofilowej do związków karbonylowych jest reakcja ze związkami Grignarda. Reakcja ta służy jako dogodna metoda otrzymywania alkoholi. Przebieg tej reakcji jest zgodny z rozkładem ładunku w grupie karbonylowej, natomiast mechanizm tej reakcji jest złożony i nie do końca poznany:
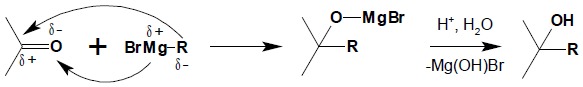
Należy zwrócić uwagę na rozkład ładunku w związkach Grignarda – reszta MgX jest zawsze spolaryzowana dodatnio natomiast atom węgla grupy alkilowej czy arylowej – ujemnie. Bezpośrednio w reakcji związku karbonylowego ze związkiem Grignarda powstaje alkoholan, który poddaje się hydrolizie przy użyciu rozcieńczonego kwasu. Odpowiednio dobierając zestaw substratów otrzymać można alkohole I-, II- i IIIrzędowe.
9. Reakcje redoks związków karbonylowych.
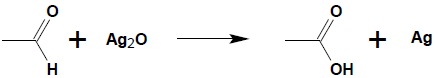
Aldehydy utleniają się bardzo łatwo do kwasów karboksylowych. Dobór środków utleniających jest w dużej mierze dowolny, ponieważ aldehydy można utleniać zarówno przy użyciu silnych środków utleniających (chromiany(VI), manganian(VII)) – powstające w wyniku reakcji kwasy są odporne na ich działanie, jak i bardzo łagodnie działających utleniaczy takich jak srebro(I) – reakcja lustra srebrnego, czy miedź(II):
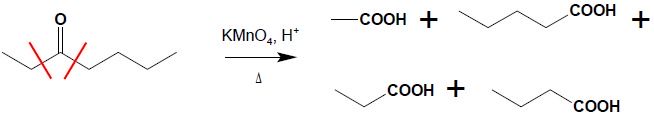
Utlenienie ketonów możliwe jest jedynie z rozpadem szkieletu węglowego cząsteczki, przebiega zatem bardzo trudno, w bardzo ostrych warunkach. W wyniku reakcji następuje rozpad wiązania C-C po obu stronach grupy karbonylowej i powstaje mieszanina kwasów karboksylowych:
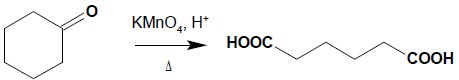
Ze względu na ostre warunki reakcji i powstawanie mieszanin produktów reakcja ta nie ma znaczenia praktycznego. Wyjątkiem jest utlenianie ketonów cyklicznych – powstają wtedy kwasy dikarboksylowe:
Zarówno aldehydy jak i ketony można stosunkowo łatwo zredukować do odpowiednich alkoholi. Najczęściej stosowane są kompleksowe wodorki – NaBH4 lub LiAlH4, przy czym NaBH4 jest stosowany zdecydowanie częściej jako łagodniejszy i bardziej selektywnie działający reduktor, który można również stosować w roztworach wodnych:
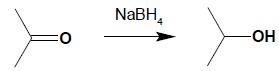
Do redukcji związków karbonylowych użyć można również gazowego wodoru w obecności katalizatora jednak należy pamiętać, że reduktor ten nie jest selektywny w swym działaniu (kompleksowe wodorki redukują jedynie grupy polarne i są bierne wobec wiązań C=C):
![]()
10. Redukcyjne usunięcie grupy karbonylowej.
Przemiana grupy karbonylowej w metylenową (C=O -> CH2) może zostać zrealizowana na dwa różne sposoby:
10.1. Redukcja metodą Clemmensena jest jednoetapowym procesem, zachodzącym w temperaturze wrzenia, pod wpływem amalgamatu cynku (roztwór cynku w rtęci) w kwasie solnym:
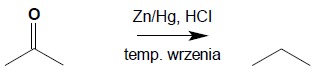
10.2. Redukcja metodą Wolffa-Kiżnera jest procesem dwuetapowym, w którym w silnie zasadowym środowisku, w wysokiej temperaturze, następuje rozkład hydrazonu aldehydu czy ketonu, otrzymanego w pierwszym etapie procesu:
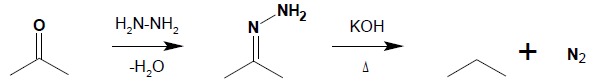
Proces ten można prowadzić bez wydzielania powstającego w pierwszym etapie hydrazonu. Często stosuje się wtedy glikole jako rozpuszczalniki, ze względu na możliwość osiągnięcia wysokiej temperatury (rzędu 200ºC) potrzebnej do termicznego rozkładu hydrazonu.
11. Reakcja Cannizzaro.
Aldehydy nieposiadające atomów wodoru w pozycji α ulegają w silnie zasadowym środowisku reakcji dysproporcjonowania, w wyniku której z dwóch cząsteczek aldehydu powstają cząsteczki kwasu i alkoholu. Reakcja rozpoczyna się od ataku nukleofilowego anionu OH– na karbonylowy atom węgla, a następnie przeniesieniu na drugą cząsteczkę aldehydu ulega anion wodorkowy:
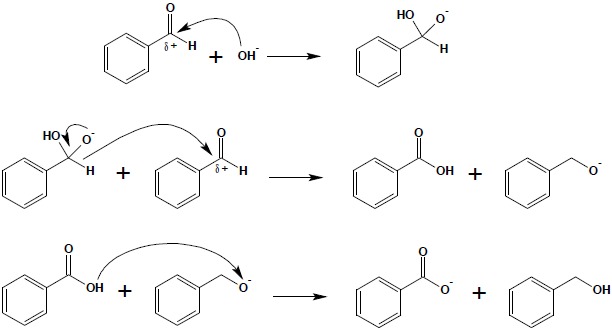
Ostatni etap reakcji polega na wymianie protonu pomiędzy kwasem a zasadą (anion alkoholanowy).
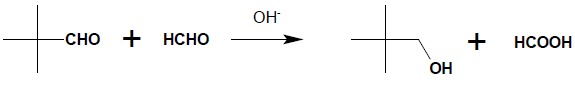
Możliwa jest również mieszana reakcja Cannizzaro, jednakże jedynym praktycznym zastosowaniem jest reakcja redukcji aldehydów za pomocą formaldehydu – bowiem jedynie w takiej kombinacji substratów przewidywalny jest kierunek reakcji (ze względu na potencjał redox formaldehyd zawsze ulega utlenieniu natomiast drugi partner reakcji – redukcji):
12. Tautomeria keto-enolowa.
W wyniku odłączenia protonu w pozycji α względem grupy karbonylowej powstaje anion, którego ładunek ujemny jest zdelokalizowany wskutek zjawiska mezomerii (dzięki temu uprzywilejowany jest taki sposób oderwania protonu od związku karbonylowego, a pozycja α względem grupy karbonylowej jest drugim – oprócz samej grupy karbonylowej, miejscem aktywnym w związkach karbonylowych). Ponowne przyłączenie protonu może zatem nastąpić przy węglu α (odtworzenie wyjściowego związku) lub przy karbonylowym atomie tlenu w wyniku czego powstaje forma enolowa:
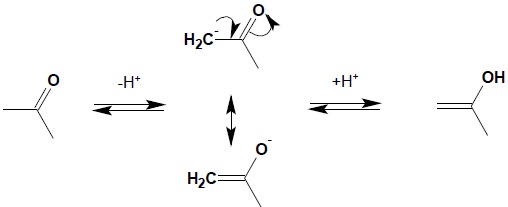
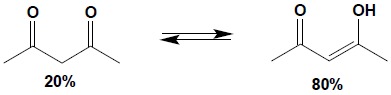
Zjawisko to nosi nazwę tautomerii keto-enolowej. Dla prostych związków karbonylowych zawartość formy enolowej w stanie równowagi jest znikoma natomiast jeżeli w wyniku powstania formy enolowej pojawia się sprzężony układ wiązań podwójnych, zawartość formy enolowej wzrasta w sposób zdecydowany:
13. Kondensacja aldolowa
Kondensacja aldolowa jest reakcją wykorzystującą reaktywność związków karbonylowych w pozycji α. Reakcję przeprowadza się w obecności zasad, powodujących w pierwszym etapie reakcji oderwanie protonu w pozycji α i otrzymanie stabilizowanego przez mezomerię (patrz wyżej) anionu:
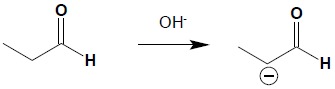
W drugim etapie reakcji następuje atak nukleofilowy anionu na karbonylowy atom węgla drugiej cząsteczki:
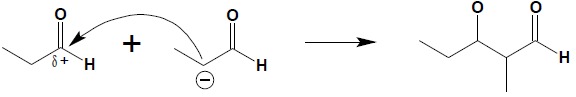
W ostatnim etapie następuje wymiana protonu pomiędzy cząsteczką wody a anionem alkoholanowym z odtworzeniem katalizatora i uzyskaniem końcowego produktu reakcji – aldolu:
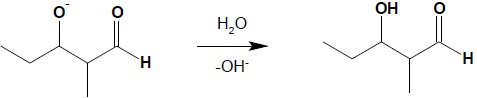
Należy zwrócić uwagę, iż odłączenie protonu, które następuje w pierwszym etapie reakcji, zachodzi jedynie w pozycji α, niezależnie od długości łańcucha, zatem do kondensacji aldolowej są zdolne jedynie takie aldehydy, które posiadają atom wodoru w pozycji α. Kondensacja aldolowa jest procesem równowagowym, jednakże dla aldehydów równowaga jest przesunięta w kierunku tworzenia się aldoli. W przypadku ketonów sytuacja jest odmienna i ze względu na bardzo małe wydajności kondensacji aldolowej ketonów w środowisku zasadowym się nie przeprowadza.
Możliwe jest wykonanie mieszanej kondensacji aldolowej, jednakże w praktyce, aby uniknąć powstawania mieszanin produktów, ogranicza się ją do przypadków, w których tylko jeden związek karbonylowy posiada atomy wodoru w pozycji α:
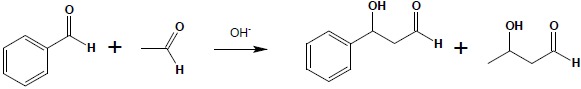
Nawet w takim przypadku powstają jednak dwa produkty – anion powstały z acetaldehydu może bowiem reagować z cząsteczką benzaldehydu jak i z drugą cząsteczką acetaldehydu. Przeprowadzając reakcję mieszanej kondensacji aldolowej, dąży się do ograniczenia ilości produktu powstałego w wyniku reakcji aldehydu, posiadającego atom wodoru w pozycji α „samego ze sobą” poprzez użycie drugiego partnera reakcji w nadmiarze. Wtedy powstały anion ma znacznie większe szanse na przereagowanie z cząsteczką drugiego związku karbonylowego i produkt kondensacji mieszanej staje się produktem głównym.
14. Reakcje addycji do nienasyconych związków karbonylowych.
W sprzężonym układzie wiązanie podwójne – grupa karbonylowa bardzo często występuje addycja typu 1,4 co prowadzi do produktów pozornie sprzecznych z regułą Markownikowa. Rozpatrując reakcję addycji HBr do aldehydu akrylowego (prop-2- enalu – najprostszego przedstawiciela α,β-nienasyconych związków karbonylowych) otrzymuje się w wyniku reakcji 3-bromopropanal zamiast spodziewanego (zgodnie z prosto pojmowaną regułą Markownikowa) 2-bromopropanalu.
W układzie sprzężonym następuje polaryzacja całego układu ze względu na obecność elektroujemnego atomu tlenu:
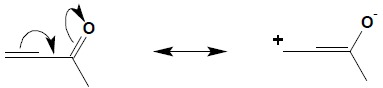
Zgodnie z rozkładem ładunku widocznym w drugiej strukturze mezomerycznej określenie kierunku addycji cząsteczki HBr staje się oczywiste: Ostatnim etapem jest proces tautomerii keto-enolowej, prowadzący do odtworzenia układu karbonylowego.
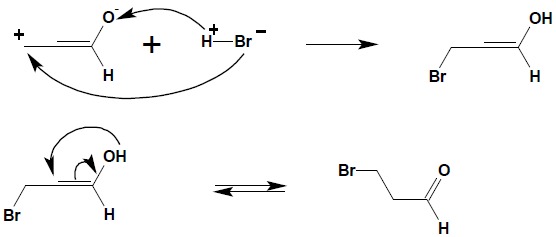
Dla substratów reagujących w reakcjach addycji z izolowaną grupą karbonylową (np.: HCN, związki Grignarda) często obserwuje się powstawanie mieszanin produktów addycji do grupy karbonylowej i addycji 1,4 do układu sprzężonego bądź możliwe jest sterowanie kierunkiem reakcji poprzez zmianę warunków prowadzenia procesu.
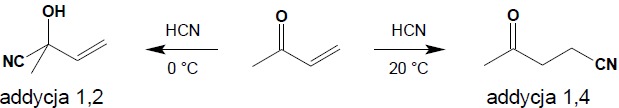
15. Otrzymywanie aldehydów i ketonów.
15.1. Utlenianie alkoholi – z alkoholi I-rzędowych otrzymuje się aldehydy, z IIrzędowych ketony
15.2. Uwodnienie alkinów.
Najprostszym przykładem (i jednocześnie jedynym, w którym powstaje aldehyd) jest reakcja Kuczerowa addycji wody do acetylenu. Reakcja jest prowadzona w obecności kwasów i soli rtęci(II) jako katalizatora:
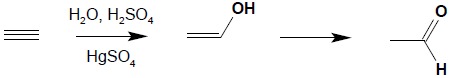
Dalsze alkiny w reakcji addycji wody dają zawsze ketony (reakcja ta przebiega zgodnie z regułą Markownikowa):
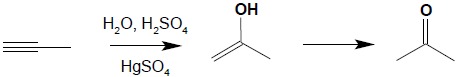
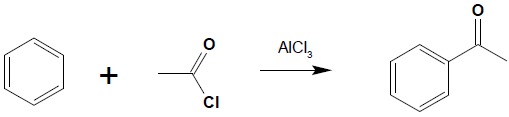
15.3. Reakcja Friedela-Craftsa
Otrzymywanie ketonów aromatyczno-alifatycznych i aromatyczno-aromatycznych z chlorków kwasowych: