
PRZEDRUK, oryginał dostępny pod adresem www
Tytuł oryginalny: Materiały dodatkowe – związki azotowe
Autor: dr Marek Żylewski
Uniwersytet Jagielloński (www)
Wydział Farmaceutyczny Collegium Medicum (www)
Katedra Chemii Organicznej (www)
Kierownik: Prof. UJ, dr hab. Marek Cegła
Adres:
ul. Medyczna 9
30-688 Kraków
Kontakt: tel. 012 620 55 00
Związki nitrowe
1. Redukcja związków nitrowych.
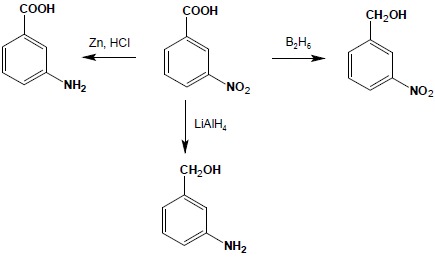
Grupa nitrowa jest bardzo podatna na działanie szeregu różnych środków redukujących. W połączeniu z łatwością otrzymywania aromatycznych związków nitrowych, otwiera to dogodną drogę syntezy pierwszorzędowych amin aromatycznych. Jako środki redukujące z powodzeniem stosować można metale – Fe, Sn, Zn, sole żelaza(II), sole cyny(II) i wodór w obecności katalizatorów. Używając wielosiarczku sodu czy amonu można przeprowadzić selektywną redukcję jednej grupy nitrowej w dinitropochodnych.:

Ze względu na to, że grupa nitrowa ulega redukcji bardzo łatwo, selektywną reakcję redukcję innych grup funkcyjnych w obecności grupy nitrowej wykonuje się bardzo rzadko. Do nielicznych środków redukujących niereagujących z grupą nitrową należy diboran – B2H6:
2. Otrzymywanie związków nitrowych
Alifatyczne związki nitrowe otrzymuje się w reakcji substytucji nukleofilowej, działając azotanem(III) sodu lub srebra na halogenopochodną:
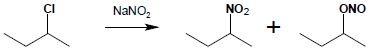
Jako produkt uboczny powstaje zawsze ester kwasu azotowego(III) – azotan(III) alkilu. Stosunek ilości produktów zależny jest od doboru substratów i warunków prowadzenia reakcji.
Aromatyczne związki nitrowe najczęściej otrzymuje się poprzez bezpośrednie nitrowanie układu aromatycznego.
Aminy
1. Zasadowość amin.
Obecność wolnej grupy elektronowej zlokalizowanej na atomie azotu amin warunkuje zasadowe właściwości amin. Im większa jest zatem gęstość elektronowa na atomie azotu, tym silniejszą zasadą powinna być odpowiednia amina. Ze względu na znaczne podobieństwo strukturalne bardzo dobrym punktem odniesienia jest amoniak. Wprowadzenie grup alkilowych, ze względu na ich elektronodonorowy charakter (porównaj wpływ skierowujący podstawników w układzie aromatycznym), powoduje zwiększenie gęstości elektronowej na atomie azotu, tym większe, im więcej grup alkilowych przyłączonych będzie do atomu azotu. Zgodnie z tym, najsilniejszymi zasadami wśród amin winny być aminy III-rzędowe i tak bywa w przypadku amin o krótkich łańcuchach węglowych. Jednakże bardzo istotnym czynnikiem wpływającym na zasadowość amin jest również łatwość solwatacji kationu powstającego z aminy, determinowana poprzez dostępność dla cząsteczek rozpuszczalnika atomu azotu. Zwiększenie liczby grup alkilowych i ich rozmiarów powoduje, ze względów przestrzennych, trudniejszy dostęp do atomu azotu, a zatem utrudnia solwatację kationu amoniowego. Konkurencyjność efektu solwatacyjnego i elektronodonorowego sprawia, iż najczęściej obserwowany dla amin alifatycznych szereg zasadowości układa się następująco: aminy II-rzędowe > aminy III-rzędowe > aminy I-rzędowe > amoniak. Różnice w zasadowości różnych amin alifatycznych nie są zbyt wielkie, natomiast należy pamiętać, iż aminy alifatyczne są zasadami silniejszymi od amoniaku.
W przypadku amin aromatycznych wprowadzenie pierścienia aromatycznego powoduje znaczący spadek zasadowości ze względu na elektronoakceptorowy charakter tego układu (wolna para elektronowa azotu amin aromatycznych ulega częściowemu uwspólnieniu z elektronami π układu aromatycznego). Aminy aromatyczne są zatem zasadami znacznie słabszymi od amoniaku i ich zasadowość spada wraz ze wzrostem liczby reszt aromatycznych przyłączonych do atomu azotu. Acylowe pochodne amin (amidy) nie wykazują własności zasadowych ze względu na silnie elektronoakceptorowy wpływ grupy karbonylowej stojącej bezpośrednio przy atomie azotu.
2. Aminy jako związki nukleofilowe.
Obecność wolnej pary elektronowej zlokalizowanej na atomie azotu warunkuje nie tylko własności zasadowe amin (jak również amoniaku), ale determinuje też silny charakter nukleofilowy tej klasy związków. Aminy, w reakcjach substytucji nukleofilowej, przewyższają reaktywnością pochodne tlenowe takie jak alkohole czy etery. Ich charakter nukleofilowy przejawia się w reakcjach z halogenopochodnymi alifatycznymi (synteza Hofmanna amin), kwasami i ich pochodnymi (synteza amidów) i związkami karbonylowymi (synteza imin i enamin).
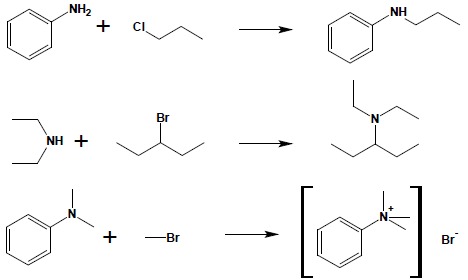
2.1. Reakcja amin (i amoniaku) z halogenopochodnymi. Alkilowanie amoniaku i amin jest najprostszą metodą syntezy amin:
![]()
W pierwszym etapie reakcji powstaje sól amoniowa, z której otrzymuje się wolną aminę pod wpływem silnej zasady nieorganicznej. W ten sposób można otrzymywać aminy wyżej rzędowe, zarówno aromatyczne, jak i alifatyczne, stosując odpowiedni dobór substratów (w równaniach pominięto etap powstawania soli):
Bardzo istotną komplikacją występującą w tej metodzie syntezy amin jest powstawanie wyżej rzędowych pochodnych. Projektując otrzymanie aminy Irzędowej w reakcji alkilowania amoniaku, zawsze otrzymuje się również dużą ilość (sięgającą 40%) aminy II-rzędowej, a także niewielką ilość aminy IIIrzędowej i IV-rzędowej soli amoniowej:
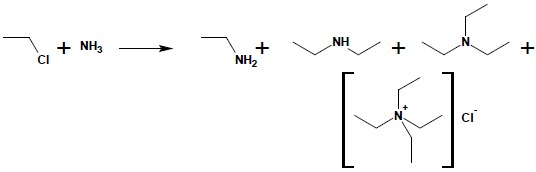
Aby zwiększyć wydajność otrzymania pożądanego produktu i zminimalizować ilość powstających wyżej rzędowych produktów w syntezie używa się dużego nadmiaru amoniaku lub aminy podlegającej reakcji alkilowania.
2.2. Reakcja amin z pochodnymi kwasów karboksylowych.
Acylowanie amin chlorkami, bezwodnikami czy estrami jest typową reakcją przebiegającą jako nukleofilowe podstawienie przy karbonylowym atomie węgla. Do reakcji używa się amoniaku, amin I- i II-rzędowych otrzymując niepodstawione i podstawione amidy:
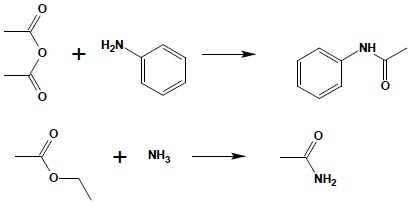
2.3. Reakcja amin ze związkami karbonylowymi
Reakcja została omówiona w części poświęconej związkom karbonylowym. Należy pamiętać, iż związki karbonylowe nie dają stabilnych produktów w reakcji z amoniakiem, z aminami I-rzędowymi dają iminy a z II-rzędowymi enaminy:
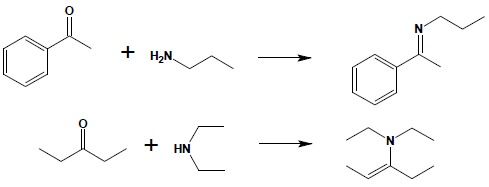
3. Utlenianie amin
Przebieg reakcji utleniania amin w sposób znaczący zależy od ich budowy chemicznej. W zależności od tego, z jaką aminą mamy do czynienia, otrzymuje się różne produkty końcowe procesu utleniania:
3.1. Utlenianie amin I-rzędowych
Aromatyczne aminy I jak i II rzędowe są generalnie bardzo wrażliwe na działanie czynników utleniających (nawet tak łagodnie działających, jak odczynnik Tollensa czy tlen z powietrza), pod których wpływem zachodzą skomplikowane procesy oksydacyjne połączone z reakcjami kondensacji w kierunku bardzo złożonych, wielopierścieniowych związków heterocyklicznych. Ciemnienie preparatów amin pod wpływem tlenu z powietrza (szczególnie w obecności światła czy śladów jonów metali ciężkich), czy też działania na aminy takimi utleniaczami jak np. chloran(VI) potasu, dichromian(VI) sodu czy chlorek żelaza(III) jest wynikiem powstawania tzw. czerni aniliniowej – jednego z najłatwiej dostępnych czarnych barwników, wytwarzanego bezpośrednio na włóknie. Silniejsze utlenianie prowadzi do otrzymania chinonów, dlatego też otrzymywanie związków nitrowych na drodze utleniania amin aromatycznych jest poważnie utrudnione i nie można w takim celu używać silnie działających środków utleniających:
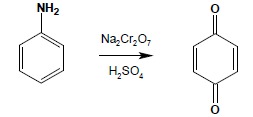
Aminy alifatyczne, jak i aromatyczne (pod warunkiem użycia odpowiednich, łagodnych środków utleniających) utlenić można do związków nitrowych. Najczęściej stosowanymi utleniaczami są nadtlenek wodoru, nadkwasy, ewentualnie (w przypadku amin alifatycznych) manganian(VII) potasu:
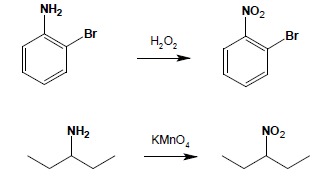
3.2. Aminy II-rzędowe utlenić można w bardzo łagodnych warunkach do pochodnych hydroksyloaminy, które jednak bardzo łatwo utleniają się dalej w kierunku powstawania złożonych mieszanin produktów:
![]()
3.3. Aminy III-rzędowe utleniają się z utworzeniem N-tlenków, które w przypadku amin alifatycznych służą jako metoda przekształcenia amin w alkeny na drodze eliminacji grupy aminowej
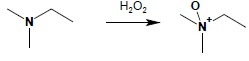
4. Reakcja amin z kwasem azotowym(III)
Kwas azotowy(III) nie istnieje w stanie wolnym. Otrzymuje się go bezpośrednio w mieszaninie reakcyjnej poprzez zakwaszenie azotanów(III) np. kwasem solnym. Reakcję najczęściej prowadzi się w temperaturze ok. 0°C (lub nawet niższej). Wykorzystywana jest zarówno w celach preparatywnych, jak i analitycznych. Pierwszy, uchwytny produkt reakcji jest wspólny dla wszystkich typów amin – w wyniku odwracalnej reakcji aminy z najprawdopodobniej powstającym w różnych przemianach równowagowych N2O3 powstaje kation N-nitrozoamoniowy:
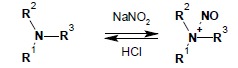
Dalsze losy powstałego kationu zalezą od budowy aminy:
4.1. Aminy III-rzędowe nie wykazują żadnej dalszej reakcji, wobec tego ze względu na odwracalny charakter powyższej reakcji uważa się je za odporne na działanie kwasu azotowego(III). Wyjątkiem są tu III-rzędowe aminy aromatyczne, nieposiadające żadnych podstawników dezaktywujących w pierścieniu aromatycznym. W takim przypadku może zachodzić reakcja substytucji elektrofilowej do pierścienia z utworzeniem brunatnych nitrozopochodnych – jednakże nie jest to reakcja przebiegająca w grupie aminowej:
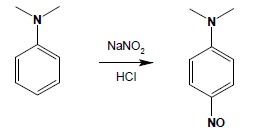
4.2. Aminy drugorzędowe tracą proton, przechodząc w N-nitrozoaminy o charakterystycznym żółto-brunatnym zabarwieniu i oleistej konsystencji:
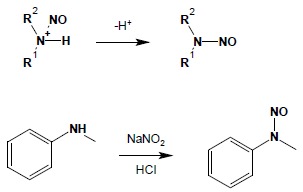
4.3. Aminy I-rzędowe tracą cząsteczkę wody z utworzeniem kationów diazoniowych.
Alifatyczne kationy diazoniowe ulegają natychmiastowemu rozkładowi z wydzieleniem cząsteczki azotu i powstaniem reaktywnego karbokationu, reagującego ze składnikami mieszaniny, co w rezultacie prowadzi do otrzymania złożonej mieszaniny produktów. Aromatyczne kationy diazoniowe są natomiast trwałe w niskich temperaturach i mogą zostać, bez wyodrębniania, użyte do dalszych reakcji chemicznych:
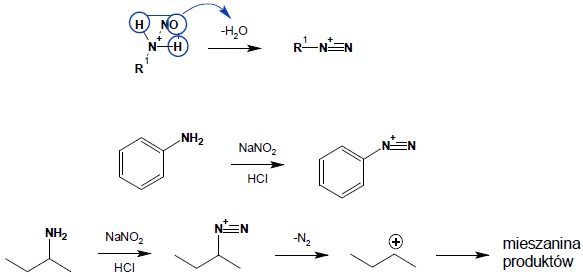
5. Metody otrzymywania amin
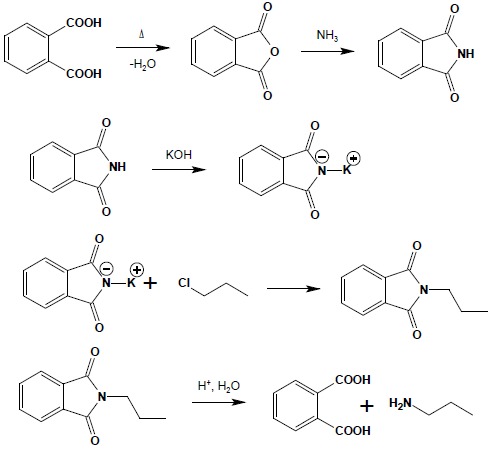
5.1. Synteza Gabriela
Ze względu na ograniczenia w bezpośredniej syntezie amin I-rzędowych metodą Hofmanna (powstawania jako produktów ubocznych wyżej rzędowych amin) opracowano metody pozbawione tych wad. W syntezie Gabriela wykorzystuje się kwasowy charakter protonów amidowych i polega ona na alkilowaniu soli sodowych lub potasowych ftalimidu (który otrzymać można w dwuetapowej syntezie z kwasu ftalowego), a następnie hydrolizie tak otrzymanej N-alkilowej pochodnej ftalimidu:
5.2. Redukcja związków nitrowych
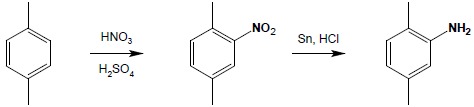
Otrzymywanie amin I-rzędowych w wyniku redukcji związków nitrowych jest bardzo dogodną metodą syntezy, o ile są dostępne odpowiednie substraty do reakcji. Ze względu na łatwość w otrzymywaniu aromatycznych związków nitrowych metodę tą stosuje się głównie do otrzymywania amin aromatycznych:
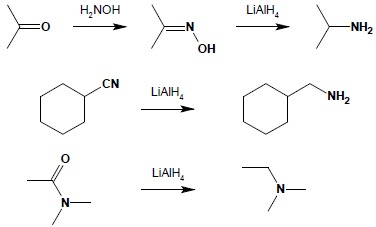
5.3. Redukcja oksymów związków karbonylowych, nitryli i amidów.
Reakcje redukcji wymienionych połączeń pozwalają na otrzymanie odpowiednich amin z dobrymi wydajnościami:
5.4. Degradacja Hofmanna
Reakcja Degradacji Hofmanna pozwala na otrzymanie amin I-rzędowych o jeden atom węgla krótszych od wyjściowych amidów. Substratem w tej reakcji jest niepodstawiony amid kwasu karboksylowego, na który działa się chloranami(I) lub bromianami(I), często otrzymywanymi bezpośrednio w mieszaninie reakcyjnej poprzez np. wkraplanie wody bromowej do roztworu NaOH (2 NaOH + Br2 = NaBrO + NaBr + H2O. Reakcja biegnie poprzez stadium N-bromoamidu, który ulega przegrupowaniu do izocyjanianu, z wydzieleniem anionu bromkowego:

Powstający izocyjanian natychmiast hydrolizuje do aminy I-rzędowej:
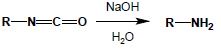
Reakcja doskonale nadaje się do syntezy amin pierwszorzędowych zarówno alifatycznych, jak i aromatycznych:
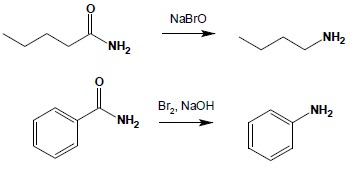
5.5. Reakcja Mannicha
Reakcja ta służy najczęściej do otrzymywania amin III-rzędowych poprzez łączenie fragmentów węglowodorowych. Substratami są w niej najlepiej aminy IIrzędowe (choć możliwe jest wykonanie na aminach I-rzędowych. lecz ze względu na możliwe komplikacje unika się stosowania takich amin), formaldehyd, jako fragment łącznikowy oraz związek posiadający czynne atomy wodoru – mogą to być związki karbonylowe: aldehydy, ketony, estry, najlepiej posiadające ugrupowanie C-H pomiędzy dwoma grupami karbonylowymi. Reakcja biegnie według następującego schematu:
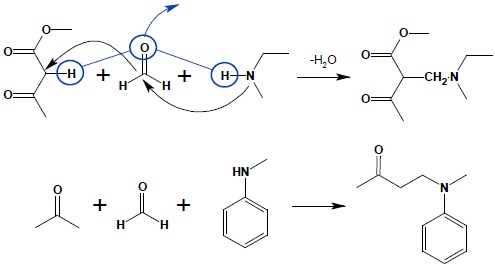
Aromatyczne sole diazoniowe
1. Reaktywność kationów diazoniowych
Sole diazoniowe powstają w wyniku reakcji amin I-rzędowych z HNO2 (patrz wyżej). Alifatyczne sole diazoniowe są bardzo nietrwałe i ulegają natychmiastowemu rozkładowi z wydzieleniem cząsteczki azotu. Aromatyczne kationy w niskiej temperaturze (nie więcej niż 5ºC) są dosyć stabilne, pozwalając na szerokie ich wykorzystanie do kolejnych syntez. Reakcje aromatycznych kationów diazoniowych można podzielić na reakcje przebiegające bez wydzielenia azotu (oba atomy azotu pozostają w produkcie reakcji) oraz z wydzieleniem azotu (w miejsce atomów azotu kationu diazoniowego wchodzi nowy podstawnik).
2. Redukcja soli diazoniowych.
Różne reduktory (np. Zn w kwasie octowym, Sn(II), NaHSO3) redukują związki diazoniowe do pochodnych hydrazyny:

3. Reakcja sprzęgania.
Niedobór elektronów w kationie diazoniowym powoduje, iż związki te mają charakter elektrofilowy. Najważniejszą reakcją, w której uwidacznia się elektrofilowy charakter soli diazoniowych, jest reakcja sprzęgania, będąca substytucją elektrofilową kationu diazoniowego do układu aromatycznego. Z racji, że sole diazoniowe są bardzo słabymi elektrofilami, reakcja sprzęgania zachodzi wyłącznie do uaktywnionych przez obecność silnie elektronodonorowego podstawnika układów aromatycznych. Substratami w reakcji sprzęgania mogą zatem być aminy aromatyczne i fenole. Produktami reakcji sprzęgania są związki azowe. W przypadku niesymetrycznie podstawionych amin i fenoli reakcja sprzęgania zawsze zachodzi zgodnie z wpływem skierowującym grupy hydroksylowej lub aminowej substratu:

Szybkość reakcji sprzęgania zależy od pH, w jakim jest prowadzona reakcja. Fenole najłatwiej ulegają reakcji sprzęgania w środowisku słabo zasadowym natomiast aminy w słabo kwaśnym. Aminy I- i II-rzędowe mogą również ulegać reakcji sprzęgania w środowisku obojętnym, lecz wtedy reakcja biegnie przy atomie azotu aminy i tworzą się dość nietrwałe układy zawierające trójatomowy łańcuch atomów azotu. Pochodne te w środowisku kwaśnym ulegają samorzutnemu przegrupowaniu do odpowiednich związków azowych:
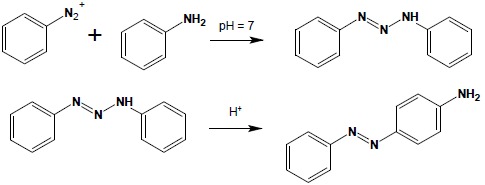
4. Otrzymywanie fenoli.
Ogrzewanie soli diazoniowych powoduje ich rozkład, w którego wyniku wydziela się azot i powstaje odpowiedni fenol. Reakcja ta, nosząca miano „reakcji zagotowania” jest jedną z lepszych metod syntezy fenoli ze względu na łagodne warunki jej przeprowadzenia:
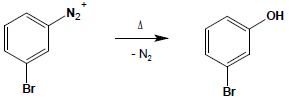
5. Otrzymywanie jodopochodnych.
Aniony jodkowe są bardzo reaktywne wobec soli diazoniowych i już na zimno, wprowadzone do roztworu soli diazoniowej powodują wydzielenie azotu i powstanie jodopochodnych aromatycznych:
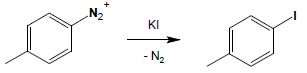
Pozostałe aniony VII grupy układu okresowego reakcji takiej nie dają.
6. Reakcja Sandmeyera
Wymiana grupy diazoniowej na atomy Cl, Br czy na grupę CN jest możliwa do zrealizowania w obecności odpowiednich soli miedzi(I) jako katalizatora:
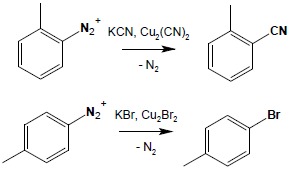
Reakcja Sandmeyera jest bardzo cenną metodą syntezy nitryli kwasów aromatycznych, z których na drodze hydrolizy można otrzymać odpowiednie kwasy.
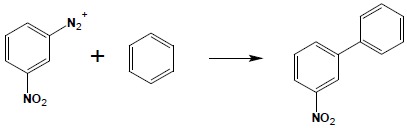
7. Reakcja Gomberga
W środowisku zasadowym związki diazoniowe reagują ze związkami aromatycznymi, dając pochodne bifenylu. Reakcja biegnie według mechanizmu rodnikowego, zatem wpływ skierowujący podstawników obecnych w układzie aromatycznym nie ma tu znaczenia:
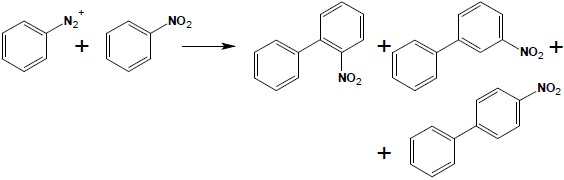
Aby uniknąć powstawania mieszanin produktów, należy tak dobierać substraty, aby podstawniki znajdowały się w cząsteczce soli diazoniowej:
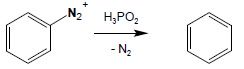
8. Redukcyjne usunięcie kationu diazoniowego.
Usunięcie grupy diazoniowej jest możliwe pod wpływem różnych reduktorów, jednakże najlepsze rezultaty otrzymuje się, działając na sól diazoniową kwasem fosfinowym:
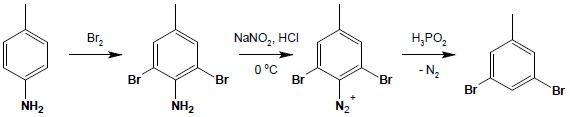
Reakcja ta ma bardzo duże znaczenie w preparatyce organicznej związków aromatycznych, pozwalając na wykorzystanie wpływu aktywującego i skierowującego grupy aminowej a później na jej łagodne usunięcie:
Amidy
1 Reaktywność amidów.
Bezpośrednie sąsiedztwo atomu azotu i grupy karbonylowej w reszcie amidowej daje możliwość sprzężenia pomiędzy elektronami π wiązania C=O a wolną parą elektronową atomu azotu. Układ ten obrazują poniższe struktury rezonansowe:
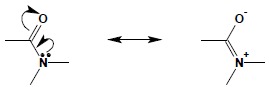
Warunkiem zaistnienia zjawiska sprzężenia jest, aby orbitale obsadzone przez elektrony biorące udział w delokalizacji mogły się na siebie bocznie nakładać, zatem atomy N, C, O przyjmują hybrydyzację sp2. Konsekwencją jest, iż wszystkie atomy budujące grupę amidową (N, C, O oraz sąsiadujące z nimi atomy) leżą na jednej płaszczyźnie. Płaska budowa wiązania amidowego ma kolosalne znaczenie dla struktury przestrzennej peptydów i białek, gdzie wiązanie to jest podstawowym fragmentem struktury spajającym poszczególne aminokwasy w łańcuch polipeptydowy. Pojawianie się wiązania podwójnego C=N w drugiej strukturze rezonansowej wiązania amidowego pokazuje również, iż amidy są najmniej reaktywnymi pochodnymi kwasów – hydroliza amidów jest jedyną reakcją przebiegającą jako podstawienie przy karbonylowym atomie węgla. Jednocześnie uwspólnienie wolnej pary elektronowej atomu azotu wyjaśnia brak właściwości zasadowych amidów.
Pomimo iż amidy są najmniej reaktywnymi pochodnymi kwasów, ulegają jednak rożnym reakcjom, których większość została już przedstawiona.
1.1 Hydroliza
Możliwa jest hydroliza amidów zarówno pod wpływem kwasów, jak i zasad. W obu przypadkach reakcja ta jest reakcją nieodwracalną:
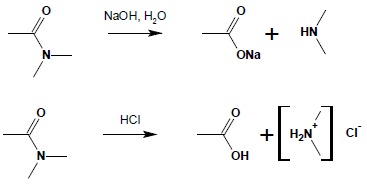
1.2 Dehydratacja
Działanie na niepodstawione amidy środkami odwadniającymi w podwyższonej temperaturze pozwala na otrzymanie nitryli:
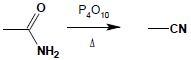
1.3 Redukcja, degradacja Hofmanna i synteza Gabriela zostały omówione w części poświęconej otrzymywaniu amin.
2 Otrzymywanie amidów.
Reakcja chlorków, bezwodników, estrów z amoniakiem bądź aminami I- lub II-rzędowymi.