
PRZEDRUK, oryginał dostępny pod adresem www
Tytuł oryginalny: Węglowodory
Autor: dr Marek Żylewski
Uniwersytet Jagielloński (www)
Wydział Farmaceutyczny Collegium Medicum (www)
Katedra Chemii Organicznej (www)
Kierownik: Prof. UJ, dr hab. Marek Cegła
Adres:
ul. Medyczna 9
30-688 Kraków
Kontakt: tel. 012 620 55 00
Alkany
1. Reakcja substytucji rodnikowej – trwałość wolnych rodników:
Reakcja substytucji rodnikowej przebiega według następującego mechanizmu:
1.1. etap I – rozpad pod wpływem światła (lub inicjatorów reakcji rodnikowych jak np. nadtlenki ROO·) cząsteczki fluorowca:
![]()
1.2. etap II – atak atomu fluorowca na cząsteczkę węglowodoru:
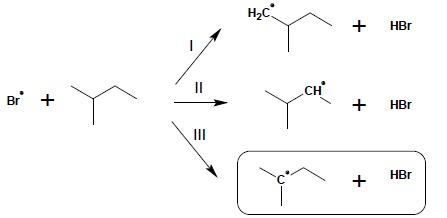
w przypadku rozgałęzionego alkanu występuje szereg miejsc, w których może nastąpić atak rodnikowy. Skutkuje to możliwością utworzenia rodników o różnej rzędowości – w powyższym przykładzie powstać mogą rodniki: pierwszorzędowy (reakcja I), drugorzędowy (reakcja II) oraz trzeciorzędowy (reakcja III). 1.3. etap III – powstały w etapie II rodnik reaguje z kolejną cząsteczką fluorowca, tworząc halogenopochodną i odtwarzając rodnikowy atom fluorowca:
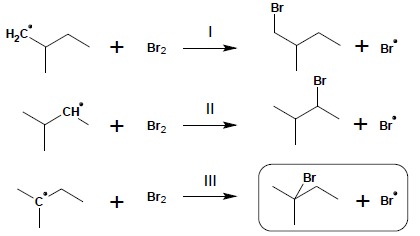
W toku reakcji bromowania otrzymuje się praktycznie wyłącznie produkt reakcji III (obwiedziony ramką), pozostałe pochodne powstają w znikomym stopniu. Przyczyna takiego wyniku reakcji bromowania leży w trwałości powstających w etapie II, jako produkt pośredni, rodników. Regułą jest, iż zawsze najchętniej tworzy się produkt najbardziej trwały, zatem powstający w reakcji III rodnik IIIrzędowy jest rodnikiem najtrwalszym. Trwałość takich rodników jest wynikiem możliwości częściowego rozproszenia ładunku niesparowanego elektronu na orbitale tworzące sąsiadujące wiązania chemiczne. Rodnikowy atom węgla posiada hybrydyzację sp2. Niesparowany elektron obsadza niezhybrydyzowany orbital typu p, prostopadły do płaszczyzny wyznaczanej przez hybrydy sp2:

Orbital ten może częściowo, bocznie nakładać się na orbitale sąsiednich wiązań typu σ pozwalając na częściową delokalizację (rozproszenie) ładunku niesparowanego elektronu (zjawisko to nosi nazwę hiperkoniugacji), jednak aby to zjawisko było możliwe, w sąsiedztwie muszą znajdować się atomy C o hybrydyzacji sp3. Im więcej takich atomów sąsiaduje z rodnikowym atomem węgla (a więc im jest on wyżej rzędowy) tym to rozproszenie jest większe a układ stabilniejszy – zatem szereg trwałości wolnych rodników przedstawia się następująco: III-rzędowy > II-rzędowy > I-rzędowy.
Ze względu na dość niską reaktywność bromu reakcja bromowana jest bardzo selektywna w doborze pozycji bromowania i w obecności III-rzędowych atomów węgla biegnie wyłącznie w kierunku podstawienia w tej pozycji. W przypadku chlorowania, ze względu na znacznie większą reaktywność tego pierwiastka, obserwuje się powstawanie mieszanin wszystkich możliwych izomerycznych pochodnych.
2. Otrzymywanie alkanów
2.1. Reakcja Würtza – reakcja halogenopochodnych alifatycznych z sodem:
2 R-Cl + 2 Na → R-R + 2 NaCl np.:
2 CH3-CH2-Cl + 2 Na → CH3-CH2-CH2-CH3 + 2 NaCl
Krzyżowej reakcji Würtza nie stosuje się ze względu na powstawanie trudnych do rozdzielenia mieszanin produktów.
2.2. Uwodornienie – katalityczna (stosowane jako katalizator są Pd, Pt, Ni) addycja wodoru do węglowodorów nienasyconych (alkenów, alkinów):
R-CH=CH-R’ + H2 → R-CH2-CH2-R’
Alkeny i alkiny
1. Addycja kwasów do wiązania podwójnego – reguła Markownikowa (trwałość karbokationów – szereg trwałości analogiczny do wolnych rodników i taka sama jego przyczyna).
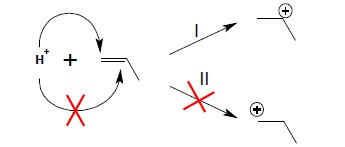
1.1. W pierwszym etapie reakcji następuje przyłączenie protonu powstałego z dysocjacji użytego do reakcji kwasu do atomu węgla stojącego przy wiązaniu podwójnym. W wyniku powstaje karbokation z ładunkiem dodatnim zlokalizowanym na sąsiednim atomie węgla. Dla niesymetrycznych alkenów reakcja biegnie zawsze w taki sposób, aby powstawał trwalszy (wyżej rzędowy) karbokation:
W dalszym etapie zachodzi przyłączenie anionu (reszty kwasowej) do karbokationu w wyniku czego otrzymuje się produkt addycji:

1.2. Najogólniej sformułowana reguła Markownikowa wskazuje, iż reakcja addycji przebiega w taki sposób, aby dodatnio naładowany fragment cząsteczki przyłączał się do mniej podstawionego (posiadającego więcej atomów wodoru) atomu węgla (powstaje wtedy trwalszy karbokation z ładunkiem zlokalizowanym na bardziej podstawionym atomie węgla). Takie sformułowanie pozwala na przewidywanie kierunku reakcji addycji do wiązania wielokrotnego cząsteczek nie odszczepiających protonów. Przykładem takiej cząsteczki jest kwas chlorowy(I) – HClO. W reakcji addycji do wiązania podwójnego zachowuje się on jak wodorotlenek:
![]()
W następnym etapie to dodatni jon Cl+ atakuje wiązanie podwójne, przejmując rolę protonu w zwykłej addycji kwasów do wiązań wielokrotnych:

Jako ostatnia ulega przyłączeniu grupa OH–:
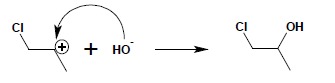
2. Wyczerpujące utlenianie alkenów przy użyciu KMnO4 w środowisku kwaśnym, na gorąco – w takich warunkach, po przejściowym utlenieniu alkenu do diolu, proces następuje dalej do rozerwania wiązania C-C. W zależności od położenia wiązania podwójnego w związku można otrzymać:
2.1. wiązanie podwójne wewnątrz łańcucha, bez rozgałęzienia:
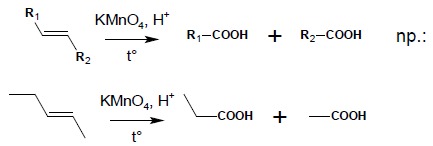
2.2. wiązanie podwójne wewnątrz łańcucha, z rozgałęzieniem:
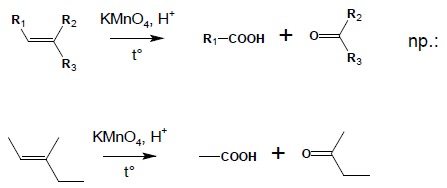
2.3. terminalne wiązanie podwójne:
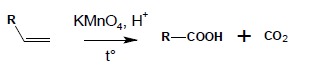
3. Łagodne utlenianie obojętnymi roztworami KMnO4 prowadzi do utworzenia wicynalnych dioli (grupy OH przy sąsiadujących atomach węgla). Reakcja biegnie poprzez etap pierścieniowego estru z resztą kwasu manganowego(VII) co powoduje określone konsekwencje stereochemiczne – wprowadzane w tej reakcji grupy OH są zawsze względem siebie w pozycji cis (reakcja ta jest reakcją stereoselektywną):
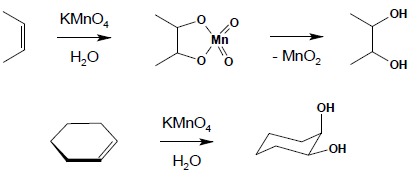
4. Ozonoliza
Reakcja ozonolizy jest specyficzną metodą utleniania alkenów prowadzącą do rozerwania wszystkich, istniejących w związku wiązań C=C z utworzeniem w ich miejscu grup karbonylowych odpowiednich ketonów lub aldehydów. Pierwszym etapem tej reakcji jest addycja cząsteczki ozonu do wiązania podwójnego z utworzeniem tzw. molozonku:

Produkt ten ulega samorzutnemu przegrupowaniu do ozonku:

Ozonek poddaje się hydrolizie w obecności środków redukujących (najczęściej w obecności Zn). Rozpad cząsteczki ozonku prowadzi do powstania odpowiednich związków karbonylowych:
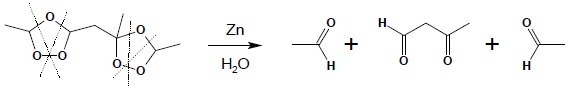
Przykład powyższy wskazuje, iż w wyniku ozonolizy powstają zazwyczaj mieszaniny produktów. Z tego powodu reakcja ta rzadko znajduje zastosowanie w syntezie (jedynie w przypadku utleniania symetrycznych alkenów) natomiast wykorzystuje się ją w celach analitycznych. Strukturę chemiczną powstających w wyniku ozonolizy związków karbonylowych można łatwo przewidzieć bez uciekania się do mechanizmu tej reakcji – należy pamiętać, iż każde wiązanie podwójne ulega rozerwaniu, a w jego miejsce pojawia się karbonylowy atom tlenu:
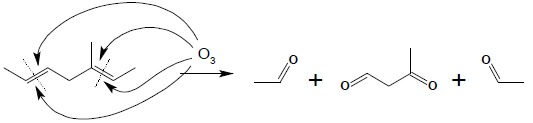
5. Sprzężony układ wiązań podwójnych – delokalizacja układu π-elektronowego
W układzie wiązanie podwójne – wiązanie pojedyncze – wiązanie podwójne (jak również w wielu innych przypadkach – ten podany jest jako najprostszy) następuje uwspólnianie elektronów π tak, iż powstające orbitale zdelokalizowane obejmują cały układ sprzężony. W przypadku butadienu CH2=CH-CH=CH2 wszystkie atomy węgla mają hybrydyzację sp2. Zatem każdy z nich posiada niezhybrydyzowany orbital typu p:
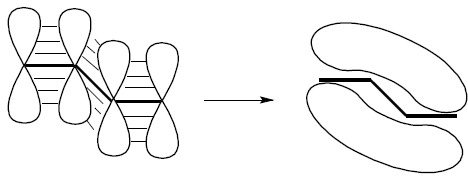
powstający orbital zdelokalizowany typu π obejmuje wszystkie cztery atomy, zatem wzór strukturalny butadienu nie odpowiada ściśle strukturze elektronowej tego związku. Dokładniejsze odwzorowanie struktury elektronowej związku zakłada wskazanie zdelokalizowanych elektronów, co zazwyczaj czynione jest za pomocą linii kropkowanej (bądź ciągłej, co przyjęło się dla układów aromatycznych) obejmującej cały układ sprzężony:
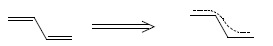
taki sposób przedstawiania wzorów strukturalnych związków jest jednak bardzo niedogodny do stosowania w praktyce, poza tym nie umożliwia on przewidywania np. biegu reakcji na podstawie możliwych położeń par elektronowych (w przykładzie przedstawionym powyżej, na podstawie wzoru pokazującego układ zdelokalizowany trudno jest przewidzieć możliwość i kierunek zachodzenia np. reakcji addycji do wiązania podwójnego). Wprowadzono zatem pojęcie struktur kanonicznych (mezomerycznych) odpowiadających granicznym, możliwym przesunięciom par elektronowych w układzie zdelokalizowanym, tak aby otrzymać odpowiadające im struktury zawierające wyłącznie wiązania zlokalizowane. Struktury te są rysowane jako zestaw i są łączone strzałką o dwóch grotach. Dla butadienu otrzymuje się zatem:
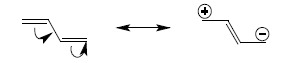
Rysując struktury kanoniczne, zakłada się, że rzeczywista budowa elektronowa jest wypadkową wszystkich możliwych struktur, natomiast z wyglądu poszczególnych wzorów można wyciągać dodatkowe informacje na temat budowy związku, których nie można przewidzieć na podstawie pojedynczego wzoru nieuwzględniającego delokalizacji (np. druga struktura kanoniczna butadienu wskazuje na obecność wiązania podwójnego pomiędzy atomami 2 i 3 zatem wiązanie między tymi atomami musi być silniejsze od zwykłego wiązania pojedynczego)
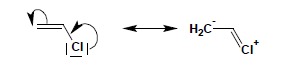
6. Reguły tworzenia struktur kanonicznych
6.1. Struktury kanoniczne można tworzyć jedynie wtedy, kiedy następuje delokalizacja elektronów obejmująca co najmniej trzy atomy – czyli kolejne trzy atomy muszą posiadać niezhybrydyzowane orbitale typu p tak aby mogły one nakładac się bocznie, tworząc zdelokalizowany orbital typu π. Zjawisko to obserwuje się dla układów typu:
6.1.1. sprzężony układ wiązań podwójnych (patrz wyżej – cztery sąsiadujące atomy lub więcej dla większych układów, posiada orbitale typu p obsadzone jednym elektronem) 6.1.2. wiązanie podwójne sprzężone z atomem posiadającym wolną parę elektronową np.:

6.1.3. wiązanie podwójne sprzężone z karbokationem (atom węgla w karbokationie ma hybrydyzację sp2, więc posiada wolny, nieobsadzony orbital typu p)

6.1.4. wiązanie podwójne sprzężone z rodnikiem (podobnie jak wyżej)
6.1.5. wiązanie podwójne sprzężone z karboanionem (podobnie jak wyżej)
6.2. Nakładanie się orbitali p jest możliwe tylko jeżeli leżą one w jednej płaszczyźnie zatem cały fragment cząsteczki, w którym ma nastąpić sprzężenie, musi być płaski.
6.3. Położenia jąder atomowych muszą być takie same we wszystkich strukturach rezonansowych.
6.4. We wszystkich strukturach musi być zachowana wartościowość pierwiastków.
6.5. Wszystkie struktury kanoniczne muszą posiadać tą samą liczbę niesparowanych elektronów (czyli przewiduje się wyłącznie przesunięcia par elektronowych, natomiast się ich nie rozdziela).
7. Addycja do układów sprzężonych
Podczas addycji fluorowców bądź kwasów do sprzężonego układu wiązań podwójnych powstaje jako etap pośredni karbokation allilowy, stabilizowany przez rezonans (patrz punkt 5.1.3) zatem znacznie trwalszy od karbokationów nawet IIIrzędowych, ale nie posiadających możliwości stabilizacji rezonansowej. W pierwszym etapie następuje zatem atak cząstki elektrofilowej kationu fluorowca lub protonu pochodzącego z dysocjacji kwasu do skrajnego atomu układu sprzężonego (bo tylko wtedy powstaje karbokation typu allilowego):
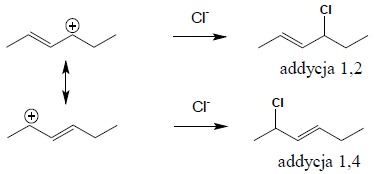
Możliwość istnienia dwóch struktur rezonansowych dla powstałego karbokationu wskazuje na dwie możliwości przyłączenia anionu kwasu, zatem pozwala przewidzieć dwa produkty reakcji addycji do układu sprzężonego:
8. Reakcja cykloaddycji 4+2 – reakcja Dielsa-Aldera
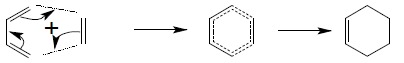
Reakcja ta należy do klasy reakcji synchronicznych, w których wszelkie przesunięcia par elektronowych powodujące zrywanie i powstawanie nowych wiązań chemicznych odbywają się w jednym etapie, bez udziału jonowych czy rodnikowych stanów pośrednich. Reakcja Dielsa-Aldera zachodzi pomiędzy sprzężonym dienem a cząsteczką tzw. dienofilu, którym może być związek posiadający wiązania wielokrotne węgiel – węgiel. Znane są również reakcje cykloaddycji, gdzie dienofilem jest cząsteczka zawierająca wiązania C=N czy N=N. Reakcja polega na jednoczesnym, kołowym przepływie elektronów w układzie sześcioczłonowym. Najprostszym przykładem jest reakcja buta-1,4-dienu z etenem:
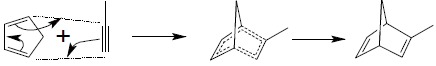
Reakcja ta z powodzeniem nadaje się do syntezy złożonych układów pierścieniowych:

Warunkiem koniecznym, aby reakcja cykloaddycji mogła zachodzić, jest możliwość występowania dienu w konformacji s-cis (single-cis) oznaczającej, iż wiązania podwójne układu sprzężonego muszą znajdować się po tej samej stronie wiązania pojedynczego, które je łączy. Dla dienów łańcuchowych warunek ten przeważnie może zostać spełniony, natomiast w przypadku dienów pierścieniowych tak być nie musi:

9. Otrzymywanie alkenów i alkinów.
Węglowodory nienasycone najczęściej otrzymuje się na drodze reakcji eliminacji. Zwykle do tego celu wykorzystuje się halogenopochodne (eliminacja HX) oraz alkohole (eliminacja H2O). W obu przypadkach kierunek reakcji eliminacji determinuje reguła Zajcewa. Zgodnie z nią w wyniku reakcji powstać powinien alken termodynamicznie trwalszy zatem jak najbardziej podstawiony przy wiązaniu podwójnym.
produkt obwiedziony ramką jest produktem głównym reakcji, lecz nie wyłącznym. Reguła Zajcewa, w przeciwieństwie do reguły Markownikowa, nie dotyczy mechanizmu reakcji, więc wskazuje jedynie na preferowany przebieg reakcji.