
Autor: Prof. dr hab. Piotr Skurski
Seminarium Pracowni Chemii Kwantowej, Warszawa, 17.02.2005
Wykład wygłoszony na Wydziale Chemii UW
PRZEDRUK, oryginał dostępny pod adresem: www
Tytuł oryginalny: Uszkodzenia struktury DNA wywoływane przez niskoenergetyczne elektrony – propozycja mechanizmu.
Zakład Chemii Kwantowej (www)
Zakład Chemii Kwantowej (ZChK) powstał 1 stycznia 2001 roku wraz z utworzeniem Katedry Chemii Teoretycznej na Wydziale Chemii UG. Obecnie w skład ZChK wchodzą pracownicy – prof. Piotr Skurski(kierownik), dr Iwona Anusiewicz i dr Sylwia Freza, a także doktoranci – mgr Celina Sikorska i mgr Iwona Świerszcz. W Zakładzie Chemii Kwantowej prace doktorskie wykonali: dr Monika Sobczyk (2004), dr Iwona Anusiewicz (2004), dr Agnieszka Sawicka (2006), dr Joanna Berdys-Kochańska (2006) i dr Sylwia Freza (2010). Obecnie w ZChK wykonywane są dwie kolejne prace doktorskie.
Od roku 1998 pracownicy Zakład Chemii Kwantowej prowadzą intensywną współpracę z grupą chemii teoretycznej w Stanach Zjednoczonych, kierowaną przez prof. Jacka Simonsa (Henry Eyring Center for Theoretical Chemistry, University of Utah, Salt Lake City).
_____________________________________________________________
Wydział Chemii Uniwersytetu Gdańskiego (www)
Efektem oddziaływania promieniowania jonizującego (pochodzącego z różnych źródeł) z komórkami organizmów żywych, jest między innymi pojawianie się tzw. elektronów wtórnych, zazwyczaj o względnie niskiej energii (od 0.1 do 20 eV). Jak wiadomo, elektrony wtórne mogą przyczyniać się do powstawania uszkodzeń DNA w organizmach żywych, a więc zarówno do pojawiania się pęknięć jednoniciowych (single-strand break, SSB), jak i dwuniciowych (double-strand break, DSB).
Wydajność, z jaką elektrony wtórne z zakresu energetycznego 1-20 eV powodują takie pęknięcia, była badana eksperymentalnie w ostatnich latach przez grupę Sanche. Jak wykazano, generowanie pęknięć odbywa się w efektywnością 8×10–4 i 2×10–4 „pęknięcia na elektron”, odpowiednio w przypadku SSB i DSB. Stwierdzono również, że wydajności te są o około 10-100 razy wyższe niż w przypadku pęknięć będących efektem oddziaływania z fotonami o znacznie wyższych energiach (sięgających nawet 25 eV).
Wykazano także, że elektrony nie muszą posiadać energii przewyższających potencjał jonizacji DNA (oszacowany na około 7.5 eV) aby wywoływać jedno- i dwuniciowe pęknięcia.
Wspomniane konkluzje były przede wszystkim efektem badań wykonanych przez Sanche i współpracowników, w ramach których suche próbki DNA (tzn. zawierające tylko tzw. wodę strukturalną w ilości około 2.5 molekuł H2O na jedną zasadę nukleinową) bombardowano wiązką niskoenergetycznych elektronów o znanej i dobrze zdefiniowanej energii kinetycznej. Po wykonaniu naświetlań próbki DNA były sprawdzane pod kątem obecności SSB i DSB. Podstawowym wnioskiem z tych eksperymentów było, jak już wspomniałem, (i) stwierdzenie możliwości wywoływania pęknięć przez elektrony o energiach 5-7 eV, czyli mniejszych niż potencjał jonizacji DNA. W szczególności wykazano, że (ii) wydajności tworzenia SSB i DSB silnie zależą od energii elektronów (nie obserwowano SSB dla energii poniżej 1 eV). Ponadto, (iii) podejrzewano, iż jednym z etapów powstawania SSB jest tworzenie stanów rezonansowych typu core-excited (o wzbudzonym rdzeniu), za czym miał przemawiać względnie długi czas życia tego typu stanów w porównaniu ze stanami shape-resonance oraz ich wysoka energia (odpowiadająca energii elektronów, dla których obserwowano większe wydajności SSB).
Wnioski sformułowane podczas tych badań eksperymentalnych sugerują zatem, iż pierwszym etapem powstawania SSB jest tworzenie rezonansowych stanów anionowych (przy czym nadmiarowy elektron zlokalizowany jest na zasadzie nukleinowej, fragmencie cukrowym bądź fosforanie – tego nie stwierdzono), natomiast etap drugi polega na rozerwaniu wiązania w komponencie cukrowym. W efekcie dochodzi do jednoniciowego pęknięcia a produkty powstałe w wyniku tego pęknięcia wchodzą w reakcje z innymi fragmentami DNA, co z kolei przyczynia się do powstawania pęknięć dwuniciowych.
Nasze wstępne badania mające na celu opracowanie przybliżonego mechanizmu dla tego procesu, rozpoczęliśmy od sprawdzenia wspomnianego schematu reakcji przy użyciu względnie prostych narzędzi kwantowochemicznych. W szczególności, rozważaliśmy możliwości wywoływania pęknięć jednoniciowych przez elektrony o energii znacznie niższej (około 1 eV). Wobec tego, naturalnym krokiem było wzięcie pod uwagę niskoenergetycznych orbitali wirtualnych w obrębie DNA. Z prac Burrowa wiadomo, że zasady nukleinowe (izolowane) tworzą anionowe stany rezonansowe, których pozycje mogłyby „pasować” do zakresu niskich energii rozważanych w eksperymentach wykonywanych przez Sanche i współpracowników. Wspomniane stany rezonansowe są jednak stanami typu shape-resonance, dlatego właśnie tego typu stany braliśmy pod uwagę w naszych obliczeniach. Wykonane przez nas obliczenia polegały więc na skanowaniu profilu energetycznego anionów tworzonych przez przyłączenie nadmiarowego elektronu do odpowiednich, antywiążących orbitali typu π. Bardziej precyzyjnie – badaliśmy ścieżkę reakcji, której współrzędna była zgodna z kierunkiem prowadzącym do powstawania SSB. Oczywiste jest, że rozmiary DNA są zbyt duże, aby można było skutecznie badać ten problem przy użyciu wysokoskorelowanych metod ab initio chemii kwantowej, czy nawet 2metody Hartree-Focka. Z uwagi na to, zdecydowaliśmy się na wyselekcjonowanie fragmentu molekularnego, który byłby reprezentatywny z punktu widzenia:
– po pierwsze – możliwości przyłączenia nadmiarowego elektronu na niskoenergetyczny orbital wirtualny π*,
– po drugie – możliwości rozerwania wiązania C–O we fragmencie cukrowym.
Tego rodzaju fragmentem struktury DNA jest pojedynczy nukleotyd. W szczególności, wybrany przez nas układ zawierał:
(i) cytozynę – jako zasadę nukleinową, (ii) fragment cukrowy, (iii) grupę fosforanową przyłączoną do fragmentu cukrowego poprzez wiązanie C–O (rozrywane przy generowaniu SSB).
Na poniższym rysunku zaznaczony jest wybrany fragment oraz wspomniane wiązanie C–O.
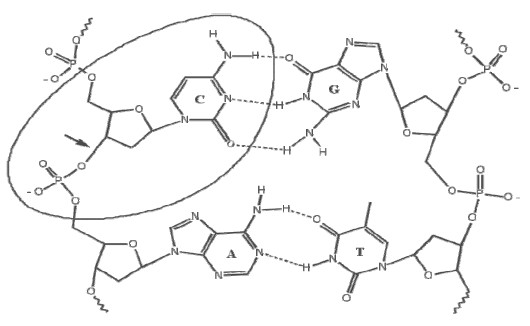
Zastosowana przez nas następnie strategia obliczeniowa była następująca:
(i) miejsca, w których dokonano sztucznego zerwania wiązań (wydzielając fragment z pełnej struktury DNA) zostały terminowane poprzez dodanie atomów wodoru. W ten sposób uniknęliśmy pojawienia się niefizycznych centrów rodnikowych, które nie występują w układzie rzeczywistym.
(ii) Zneutralizowaliśmy naładowaną ujemnie grupę fosforanową poprzez wprowadzenie dodatkowego jonu H+. Uzasadnieniem takiego postępowania jest fakt, iż w eksperymentach z grupy Sanche posługiwano się co prawda „suchym” DNA, ale zawierającym wodę strukturalną. W tego rodzaju próbkach mamy bez wątpienia do czynienia z jonami równoważącymi ujemnie naładowane reszty fosforanowe.
(iii) Wykonaliśmy serię obliczeń dla układu obojętnego (na poziomie RHF) oraz anionowego (UHF) zmieniając wartość współrzędnej reakcji R (długość wiązania C–O) od 1.40 A do 2.20 A (z krokiem 0.05 A). Obliczenia te uwzględniały w każdym przypadku optymalizację geometrii przy zamrożonej wartości współrzędnej R. Długość wiązania 1.40 A odpowiadała obszarowi równowagowemu, natomiast długość R=2.20 A wskazuje na obszar, w którym doszło już do pęknięcia SSB.
(iv) Dla mniejszych wartości parametru R nadmiarowy elektron w układzie anionowym umieszczany był na antywiążącym orbitalu π* zlokalizowanym na zasadzie nukleinowej.
(v) Obliczenia wykonywaliśmy niezależnie (w sensie uzyskiwania niezależnych profili energetycznych) dla różnych energii nadmiarowego elektronu, a więc dla 0.2, 0.3, 0.8, 1.0, 1.3 i 1.5 eV. Za chwilę wyjaśnię szczegóły techniczne tych obliczeń.
(vi) Opisane w powyższych punktach profile energetyczne wykonaliśmy dla molekuły w fazie gazowej oraz w polu rozpuszczalnika (metodą SCRF) o różnej stałej dielektrycznej, tzn. 4.9, 10.4 i 78).
Postępowanie to doprowadziło do wygenerowania przekroju przez powierzchnię energii potencjalnej (osobno dla układu obojętnego i anionowego). W przypadku anionu przekrój ten ilustruje połączenie między anionem π* a układem o rozerwanym wiązaniu C–O.
Zanim powiem o wynikach liczbowych chciałbym zwrócić uwagę, że proponowany przez nas schemat przebiegu tego procesu zakłada migrację nadmiarowego elektronu z zasady nukleinowej (gdzie elektron jest przyłączany w pierwszym etapie tworząc krótkożyjący rezonans) do reszty fosforanowej. Migracji tej towarzyszy rozerwanie wiązania C–O i powstanie jednoniciowego pęknięcia. Oczywiście w procesie tym powstaje również centrum rodnikowe, które może w dalszym etapie być powodem powstawania kolejnych uszkodzeń w najbliższym sąsiedztwie.
Na poniższych rysunkach prezentuję sytuację początkową i końcowa. Z lewej strony widać orbital molekularny π* zasady nukleinowej, na którym zlokalizowany jest elektron po przyłączeniu do nukleotydu. Z prawej strony, natomiast, widać orbital docelowy, opisujący nadmiarowy elektron po rozerwaniu wiązania C–O.
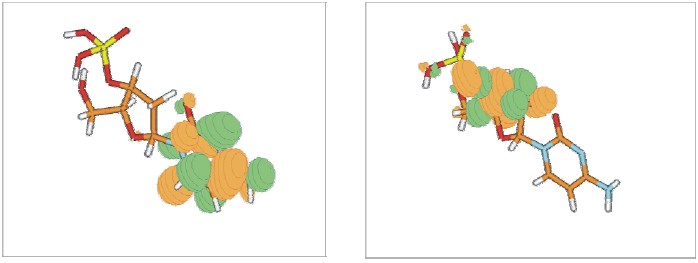
Dosyć ważne jest wyjaśnienie dlaczego nie rozważamy bezpośredniego przyłączenia nadmiarowego elektronu do antywiążącego orbitala σ* wiązania C–O. Oczywiście, tego rodzaju mechanizm jest znany, natomiast nie może on dotyczyć sytuacji, gdy SSB są indukowane przez niskoenergetyczne elektrony. Wynika to z faktu, że energia tych elektronów jest za mała, aby orbital σ* wiązania C–O mógł zostać osiągnięty, co jest spowodowane tym, że energia σ* wiązania C–O jest wysoka dla geometrii równowagowej nukleotydu. Orbital ten jest więc nieosiągalny dla elektronów wtórnych o energiach 0-2 eV, czyli takich, jakie rozważamy w naszych symulacjach. Pomimo, iż orbital σ* wiązania C–O nie jest osiągany natychmiast po przyłączeniu elektronu, uczestniczy on w omawianym procesie transferu elektronu na etapie późniejszym, dla większych długości wiązania C–O, gdzie jego energia jest już dużo niższa.
Tak jak wspomniałem, nasze profile energetyczne uzyskiwaliśmy dla różnych energii nadmiarowego elektronu w zakresie 0.2–1.5 eV. Fakt, że tworzony w pierwszym etapie stan rezonansowy anionu π* jest metastabilny, utrudnia wykonywanie tego rodzaju obliczeń. Wykorzystaliśmy tę okoliczność, jak i brak zupełności używanej bazy funkcyjnej, jako technikę umożliwiającą nam sterowanie energią nadmiarowych elektronów (spełniających w naszych obliczeniach rolę elektronów wtórnych). W szczególności, wykonywaliśmy takie skalowanie bazy, aby różnica energetyczna między obliczanym stanem anionowym a macierzystym układem obojętnym była równa symulowanej energii kinetycznej zbliżającego się elektronu. Skalowaniu podlegały wykładniki orbitali o symetriach s i p, które wnoszą dominujący wkład do orbitali antywiążących π* zlokalizowanych na zasadzie nukleinowej (cytozynie). Po przeprowadzeniu skalowania bazy (na uzyskanie odpowiedniej energii) dla geometrii równowagowej nukleotydu, stosowaliśmy tę bazę konsekwentnie skanując dany profil energetyczny reakcji. Warto również zwrócić uwagę, iż wybór wartości energii (a więc 0.2, 0.3, 0.8 eV, itd.) nie był przypadkowy lecz odpowiadał eksperymentalnej pozycji rezonansu określonej dla izolowanej cytozyny przez Burrowa i współpracowników. Dokładniej – wspomniane wartości energii charakteryzowały i uwzględniały zarówno pozycję tego rezonansu, jak i jego szerokość.
Wyniki – profile energetyczne
Uzyskane przez nas profile energetyczne (dla różnych wartości energii oraz dla różnych stałych dielektrycznych) są pokazane na poniższym rysunku:
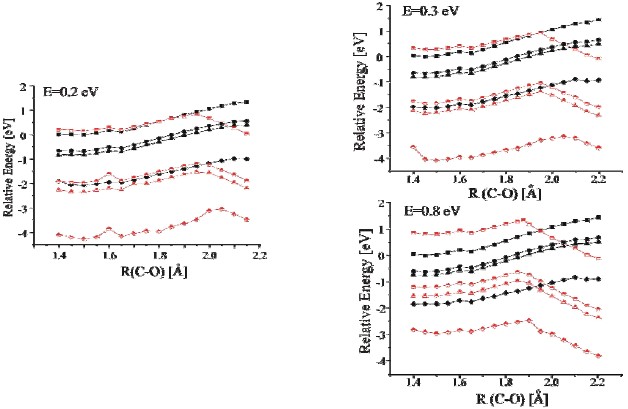
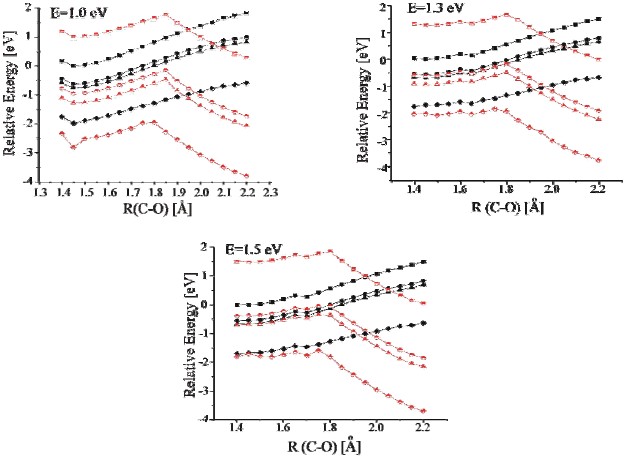
Na rysunkach pokazano również profile energetyczne odpowiednich (macierzystych) układów obojętnych w celu zilustrowania rejonów stabilności elektronowej opisywanych anionów.
Jak widać, w nieobecności efektów rozpuszczalnikowych (ε=1.0) zerwanie wiązania C–O wymaga pokonania bariery kinetycznej o wysokości 8-16 kcal/mol (w zależności od energii elektronów) ale cały proces jest egzotermiczny we wszystkich przypadkach, co jest niewątpliwie efektem dużego powinowactwa do nadmiarowego elektronu wykazywanego przez fragment fosforanowy. Można również zauważyć, że tylko w nieobecności rozpuszczalnika mamy do czynienia z metastabilnym stanem anionowym – we wszystkich pozostałych przypadkach natomiast, badane aniony są stabilne elektronowo w całym zakresie badanych długości R.
Sformułowanie dalszych wniosków jest prostsze po zebraniu tych samych wyników w tabeli:
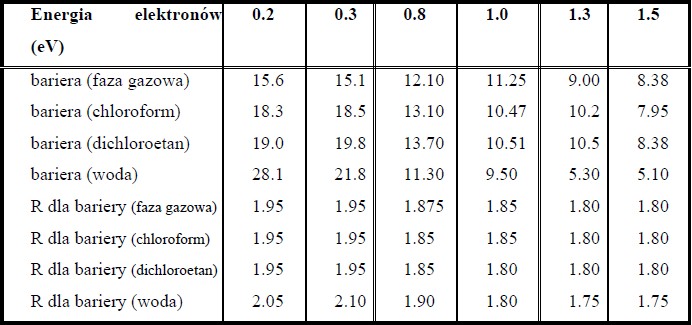
Analizując dane zawarte w powyższej tabeli można sformułować następujące wnioski:
(i) bariery kinetyczne występują dla podobnych wartości R (długość wiązania C–O), czyli w rejonie R=1.75-2.10 A, przy czym bariery występują dla nieco krótszych odległości C–O w sytuacjach, gdy energia elektronów wzrasta;
(ii) biorąc pod uwagę wszystkie rozpuszczalniki, wysokości barier zawierają się w przedziale 5-28 kcal/mol, przy czym wysokość bariery maleje wraz ze wzrostem energii elektronów, co wiąże się pośrednio z tym, że dla wyższych wartości energii mamy niejako więcej energii zgromadzonej w układzie anionowym, co niewątpliwie ułatwia pokonanie bariery kinetycznej;
(iii) dla mniejszych wartości energii elektronów bariera zazwyczaj zwiększa się wraz ze wzrostem stałej dielektrycznej, natomiast dla większych energii elektronów obserwowana jest tendencja odwrotna.
Wyniki – szybkość powstawania SSB
Na podstawie uzyskanych wyników można też dokonać oszacowania szybkości z jaką może dochodzić do pokonania bariery i generowania pojedynczych pęknięć (SSB). W tym celu wystarczy rozważyć drgania wiązania C–O, w przypadku którego wibracje zachodzą z szybkością około 3×10 13 s -1. Jeżeli pomnożymy tę wielkość przez prawdopodobieństwo określające możliwość pokonania odpowiedniej bariery kinetycznej wskutek rozciągania wiązania C–O (dla temperatury 298 K) uzyskamy następujące szybkości powstawania SSB:
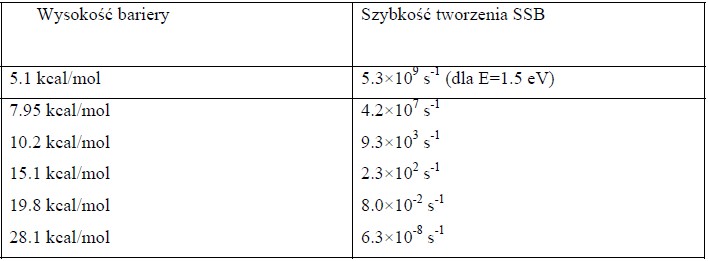
Jak pokażę za chwilę, szybkości te są mniejsze niż szybkość, z jaką elektron odbywa podróż z zasady nukleinowej do grupy fosforanowej, a więc to właśnie te szybkości będą determinować szybkość powstawania SSB (oczywiście wniosek ten dotyczy wyłącznie sytuacji i warunków, w których proponowany mechanizm jest realizowany).
Warto w tym miejscu zauważyć, że czas życia stanu rezonansowego π* dla izolowanej cytozyny wynosi około 10-13 s (wobec braku solwatacji). Jednocześnie widać, że dla niesolwatowanych nukleotydów, jedynie dla energii 1.5 eV szybkość tworzenia SSB ma szanse być nie bliżej niż trzy rzędy wielkości od szybkości odrywania elektronu od stanu rezonansowego. Nie należy się zatem dziwić, że dla energii 1.5 eV (lub niższych) nie obserwowano powstawania SSB w eksperymentach Sanche i współpracowników.
W przypadku układów solwatowanych, anion jest zawsze stabilny elektronowe, wobec tego kwestii konkurencyjności procesów autooderwania elektronu i generowania SSB nie trzeba rozważać.
Dla stałych dielektrycznych wynoszących około 4, które mogą być reprezentatywne dla natywnego DNA, szybkość powstawania SSB jest określona w zakresie 100–107 s-1 (dla elektronów z zakresu 0.2–1.5 eV).
Wyniki – szybkość transferu elektronu
Jak już wspominałem, dla każdej wartości rozważanego parametru R (długość wiązania C–O) mamy do czynienia z dwoma stanami anionowymi, które musimy rozważyć w celu dokonania pełniejszego opisu badanego procesu. Pierwszy z tych dwóch stanów diabatycznych to stan π*, w którym elektron zlokalizowany jest na antywiążącym orbitalu π w rejonie zasady nukleinowej, natomiast drugi stan diabatyczny jest stanem σ* a nadmiarowy elektron opisywany jest antywiążącym orbitalem σ wiązania C–O. Stan σ* ma wysoką energię w rejonie równowagowej długości wiązania C–O a następnie jego energia maleje, natomiast w przypadku stanu π* obserwujemy tendencję odwrotną, co zostało pokazane na rysunku:
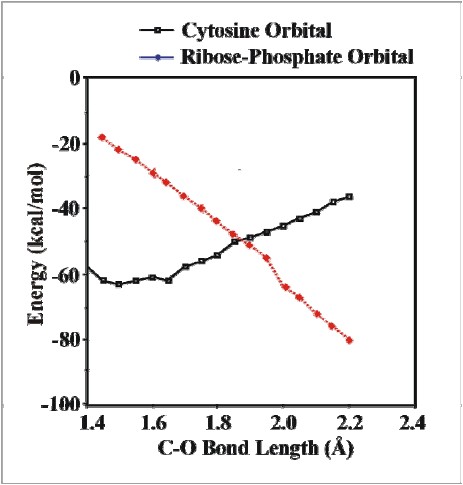
Oczywiście dla dużych wartości parametru R, układ ewoluuje do anionu fosforanowego oraz do obojętnego rodnika i to właśnie tworzenie anionu fosforanowego stanowi największą „siłę sprawczą” w procesie rozrywania wiązania C–O. Warto wspomnieć, iż grupa fosforanowa posiada wysokie powinowactwo do nadmiarowego elektronu (powyżej 4 eV), co z punktu widzenia elektronu zlokalizowanego na zasadzie nukleinowej jest perspektywą głębokiej studni potencjału, która może być osiągnięta po pokonaniu bariery kinetycznej związanej z rozerwaniem wiązania C–O.
W rejonie R=1.9 A, energia stanu σ* staje się w przybliżeniu równa energii stanu π*. W tym obszarze dochodzi do „zabronionego przecięcia” (avoided crossing), unikania się stanów w wyniku czego pojawiają się dwa stany adiabatyczne, w przypadku których region wzajemnego unikania się pokazany jest na poniższych rysunkach:
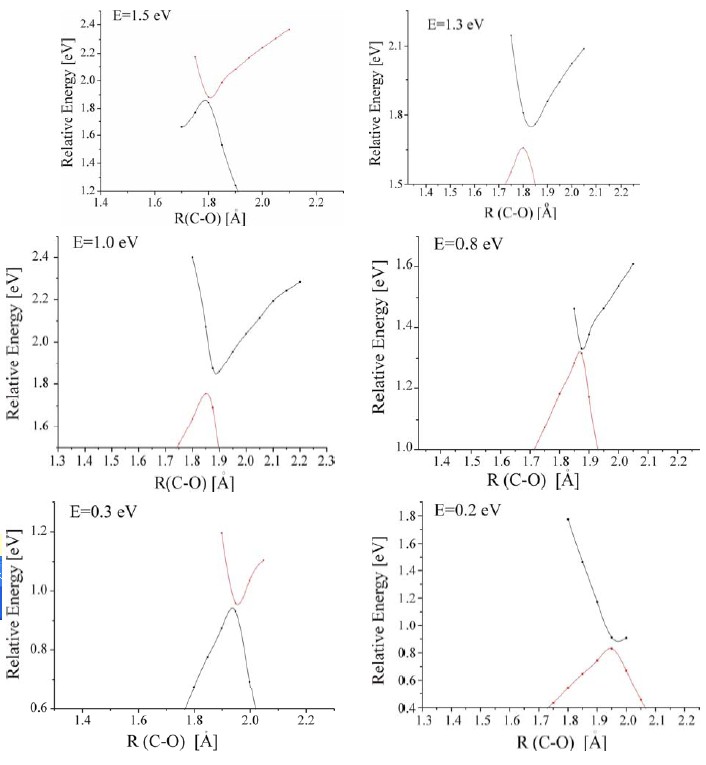
Jak widać, dla każdej z energii mamy nieco inną sytuację ale w każdym przypadku możemy oszacować „odstęp energetyczny” między krzywymi w miejscu, w którym zbliżają się one do siebie najbardziej. Umożliwia to oszacowanie szybkości, z którą nadmiarowy elektron, zlokalizowany początkowo na orbitalu π* cytozyny, przemieszcza się na orbital σ* wiązania C–O.
Odczytane z wykresów wartości energii mieszczą się w zakresie od 0.01 do 0.24 eV i odpowiadają szybkościom od 2×1012 do 6×1013 s-1.
Obliczone szybkości transferu wskazują, że nadmiarowy elektron może w rzeczywistości dosyć szybko przemieszczać się z zasady nukleinowej na fosforan. W szczególności, warto zauważyć, że uzyskane w ten sposób szybkości są dużo większe niż prędkość pokonywania barier kinetycznych (na drodze do rozerwania wiązania C–O). W związku z tym, prędkość generowania SSB (na drodze analizowanego mechanizmu) będzie zdeterminowana prędkością pokonywania bariery kinetycznej, a nie szybkością transferu elektronu w obrębie nukleotydu.
Analogiczne obliczenia wykonaliśmy również dla nukleotydu, w którym rolę zasady nukleinowej pełni tymina. Uzyskaliśmy zbliżone wyniki, co nie stanowi zaskoczenia gdyż tymina jest podobna do cytozyny nie tylko w sensie strukturalnym, ale również z punktu widzenia możliwości tworzenia stanów rezonansowych (pozycje anionowych rezonansów cytozyny i tyminy należą do najniższych spośród wszystkich zasad nukleinowych).
Zebrane dla wspomnianego układu wyniki zaprezentowane są w poniższej tabeli:
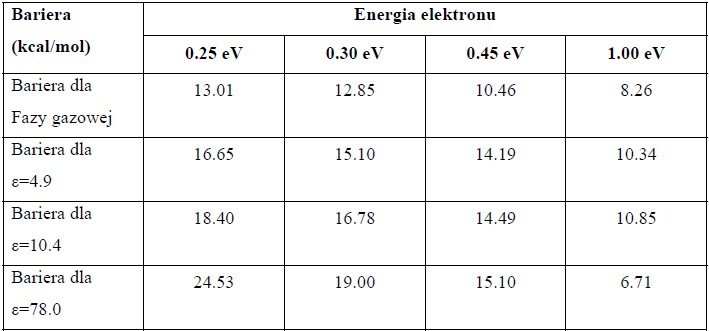
Można łatwo zauważyć, że wysokości barier zawierają się w zakresie 8-13 kcal/mol (dla fazy gazowej), przy czym wysokość 8.26 kcal/mol odpowiada energii elektronów równej 1.0 eV.
Podobnie jak dla układu zawierającego cytozynę, tylko w fazie gazowej mamy do czynienia z metastabilnym stanem anionowym, natomiast nawet słabe pole rozpuszczalnika stabilizuje tworzony anion.
Podobnie jak w przypadku nukleotydu zawierającego cytozynę, czynnikiem determinującym szybkość powstawania SSB jest tutaj wysokość bariery kinetycznej i konkurencyjność tego procesu z autoodrywaniem elektronu.
Efekty π-stacking
Kolejnym krokiem na drodze do uwzględnienia wpływu większej liczby czynników zewnętrznych na proponowany przez nas mechanizm generowania SSB w strukturze DNA, było wzięcie pod uwagę efektu związanego z obecnością innych nukleotydów. W tym celu wykonaliśmy obliczenia dla fragmentu pojedynczej nici DNA, składającego się z trzech nukleotydów (patrz poniższy rysunek):
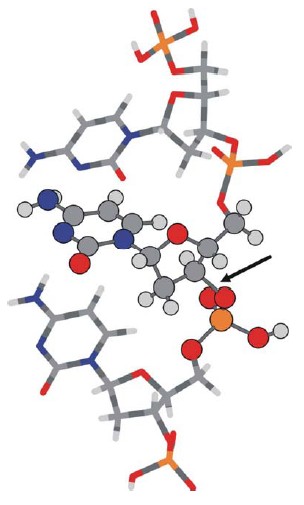
Wybrana do badań struktura pochodziła z banku danych DNA i odpowiadała fragmentowi nici złożonemu z trzech nukleotydów, z których każdy zawierał cytozynę (pełniącą rolę zasady nukleinowej). Warto wspomnieć, że sekwencja CCC stanowi ważny biologicznie kodon odpowiedzialny za kodowanie jednego z naturalnych aminokwasów – proliny.
Dla takiej struktury badaliśmy omawiany wcześniej mechanizm przeniesienia elektronu, przy czym, zgodnie z naszym założeniem a priori, rozważaliśmy zachodzący proces dla „środkowego” nukleotydu, natomiast dwa sąsiadujące układy pełniły rolę „otoczenia”. W ten sposób, znając wyniki dla izolowanego nukleotydu (również zawierającego cytozynę), można, na drodze porównania, określić wpływ bezpośredniego sąsiedztwa „zestakowanych” zasad nukleinowych na szybkość reakcji prowadzącej do zerwania wiązania C–O.
W związku z tym, że badany fragment jest w tym przypadku znacznie większy, zastosowaliśmy podejście, w ramach którego: cały układ został arbitralnie podzielony na dwie strefy: do jednej z nich – „o niskim priorytecie” – zaliczyliśmy dwa nukleotydy (górny i dolny) – dla atomów z tej strefy wykonywaliśmy obliczenia metodą HF w bazie 4-31G, natomiast dla atomów strefy drugiej („o wysokim priorytecie”), obejmującej środkowy nukleotyd, również metodą HF ale w bazie 6-311+G*. Podział na strefy jest zilustrowany na pokazanym wcześniej rysunku.
W przypadku kodonu CCC wykonaliśmy, jak dotąd, obliczenia tylko dla fazy gazowej. Optymalizacja geometrii, którą przeprowadzaliśmy dla wszystkich zamrożonych wartości parametru R, była wykonywana z więzami. Mianowicie, optymalizacji podlegały współrzędne wewnętrzne poszczególnych nukleotydów, natomiast wzajemna orientacja (ułożenie w przestrzeni) fragmentów nukleotydowych pozostawało zachowane.
Obliczenia wykonywaliśmy dla sześciu różnych wartości energii nadmiarowego elektronu (od 0.3 do 2.0 eV). Profile energetyczne, analogiczne do prezentowanych wcześniej ale w otoczeniu dwóch sąsiadujących nukleotydów, zaprezentowane są na poniższym rysunku:
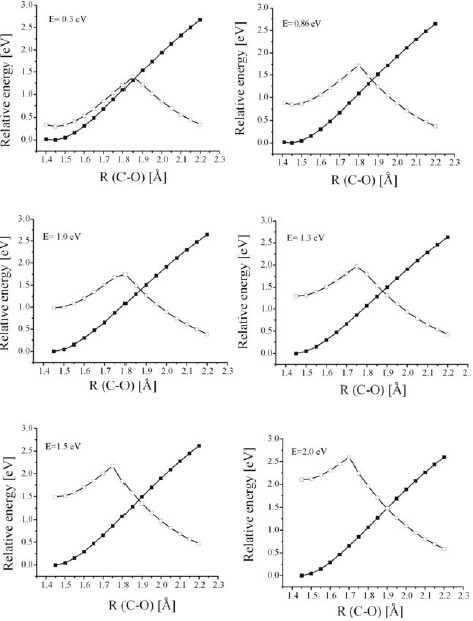
Jak wynika z tych rysunków, wysokość bariery kinetycznej zawiera się w przedziale 11-25 kcal/mol, przy czym najniższa bariera odpowiada energii elektronów równej 2.0 eV i systematycznie rośnie wraz ze spadkiem energii elektronów, poprzez 15 kcal/mol dla 1.5 eV, 18 kcal/mol dla 1.0 eV, aż do 25 kcal/mol dla 0.3 eV.
Porównanie wysokości uzyskanych w ten sposób barier z wynikami otrzymanymi uprzednio dla izolowanego nukleotydu (również w fazie gazowej) prowadzi do wniosku, iż efekty π-stacking powodują zwiększenie wysokości barier o około 8 kcal/mol. A zatem, w obecności sąsiadujących nukleotydów będziemy mieli do czynienia z redukcją szybkości tworzenia SSB na drodze rozważanego mechanizmu.
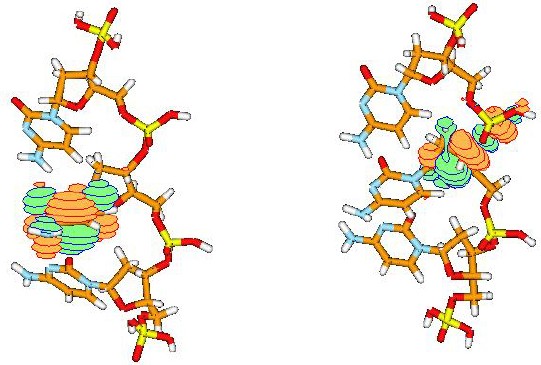
Transfer elektronu z fragmentu cytozynowego na grupę fosforanową (odpowiednie orbitale są pokazane na poniższym rysunku) jest również dla badanego kodonu znacznie szybszy niż osiąganie bariery kinetycznej, wobec czego proces ten nie jest etapem determinującym prędkość powstawania SSB.
Korzystając z wyznaczonych barier kinetycznych możemy określić odpowiadające im szybkości generowania SSB (dla różnych energii elektronu), które zostały zebrane w tabeli:
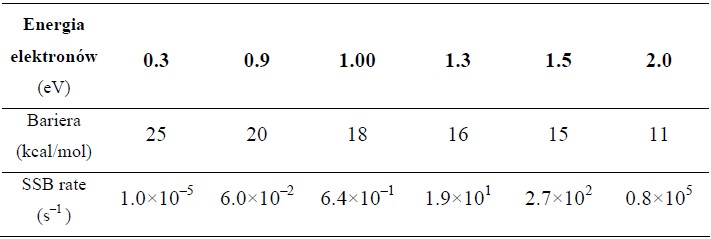
Ponieważ szybkość autoodrywania elektronu od rozważanego stanu rezonansowego wynosi około 1013 s-1, natomiast szybkość migracji elektronu na sąsiednią zasadę nukleinową (proces ten nie był rozważany w naszych pracach) wynosi 1012 s-1, widać, że prędkości tworzenia SSB zgodnie z proponowanym przez nas mechanizmem są o około (co najmniej) 8 rzędów mniejsze. Wobec tego należy stwierdzić, że tego rodzaju droga powstawania pęknięć jednoniciowych w kodonie CCC jest mało prawdopodobna dla elektronów o energiach mniejszych lub równych 2 eV, chyba, że problem niestabilności powstającego w pierwszym etapie stanu π* zostanie wyeliminowany poprzez zastosowanie polarnego, stabilizującego środowiska.
Podsumowanie
Zgodnie z postulowanym mechanizmem przewidujemy następującą sekwencję zdarzeń:
(i) Elektron posiadający energię kinetyczną w zakresie 0.2-1.5 eV przyłącza się do antywiążącego orbitala π* zlokalizowanego na zasadzie nukleinowej (elektron nie może być przyłączony do orbitala σ* wiązania C–O ze względu na jego wysoką energię odpowiadającą geometrii równowagowej nukleotydu);
(ii) Wobec braku stabilizującego otoczenia (solwatacja) utworzony rezonans może ulegać procesowi autooderwania elektronu (w ciągu około 10-13 s);
(iii) Alternatywnie, układ anionowy z elektronem przyłączonym na orbital π* może ulec geometrycznej reorganizacji (również pod wpływem otoczenia) i uzyskać stabilność elektronową (jak stwierdziliśmy, już niezbyt silne pole rozpuszczalnika umożliwia stabilizację rozważanego stanu anionowego);
(iv) Podczas drgań wiązania C–O w anionie istnieje pewne (aczkolwiek niewielkie) prawdopodobieństwo osiągnięcia krytycznej długości wiązania, dla której stany anionowe π* i σ* wykazują bardzo zbliżoną energię. Bariery kinetyczne, które muszą być pokonane na drodze do osiągnięcia takiej sytuacji zawierają się w zakresie 5-28 kcal/mol a ich wysokość silnie zależy od polarności środowiska oraz od energii elektronów wtórnych;
(v) Po osiągnięciu bariery, zlokalizowany początkowo na zasadzie nukleinowej elektron szybko się przemieszcza i powstaje stabilny elektronowe anion [(HO)2PO–O]–; oraz odpowiedni obojętny układ rodnikowy. Szybkość migracji elektronu jest znacznie większa niż szybkość osiągania bariery, stąd etap ten praktycznie nie wpływa na wydajność generowania SSB zgodnie z tym mechanizmem.
Uwagi końcowe
(i) Omawiana przez nas droga powstawania SSB w strukturze DNA dotyczy sytuacji, w której grupy fosforanowe (w sąsiedztwie zasady nukleinowej przyłączającej nadmiarowy elektron) będą zneutralizowane odpowiednimi kationami, a więc nie będą nośnikami nadmiarowego ładunku ujemnego (taka sytuacja dotyczyła niewątpliwie eksperymentów wykonywanych przez Sanche i współpracowników).
(ii) Nie sugerujemy, iż proponowany mechanizm jest realizowany w przypadku większości powstających SSB w DNA. Uważamy jednak, że może on stanowić jedną z dróg, na których realizowane są tego rodzaju uszkodzenia, a także przyczynia się do wyjaśnienia wyników eksperymentalnych dotyczących bombardowania suchych próbek DNA wiązką elektronów w niskim zakresie energii.
Podziękowania:
Dr Monika Sobczyk
Dr Iwona Anusiewicz
Mgr Joanna Berdys-Kochańska
Mgr Agnieszka Sawicka