
PRZEDRUK, oryginał dostępny pod adresem www
Tytuł oryginalny: Synteza peptydów na nośniku stałym metodą Fmoc/tBu
Uniwersytet Gdański (www)
Wydział Chemii (www)
Katedry Chemii Bioorganicznej (www)
Kierownik: Prof. dr hab. Krzysztof Rolka
Adres:
ul. Sobieskiego 18/19
80-952 Gdańsk
Kontakt: tel. 58 52 35 386
____________________________________________________________________________
Synteza peptydów metodą klasyczną, inaczej zwaną syntezą w roztworze, napotyka szereg trudności. Metoda ta jest bardzo pracochłonna i czasochłonna, ponieważ do zsyntezowania peptydu trzeba przeprowadzić szereg procesów tworzenia wiązania peptydowego, etapów usuwania osłon grup aminowych, a ponadto każdy otrzymany produkt pośredni powinien być oczyszczony i scharakteryzowany. Zwykle najtrudniejszym i najbardziej czasochłonnym elementem każdego etapu syntezy jest proces oczyszczania kolejnych fragmentów tworzonego peptydu. Dodatkowo utrudnienie stanowi obecność produktów ubocznych, często o własnościach zbliżonych do własności produktów głównych, które są trudne do oddzielenia. W 1962 roku Merrefield opracował nową strategię syntezy chemicznej peptydów i białek, która, podobnie jak biosynteza białek, przebiega w innej fazie. Metoda ta polega na tym, że pierwszy aminokwas wiąże się kowalencyjnie swą grupą karboksylową z nierozpuszczalnym polimerem, co ułatwia sączenie, a następnie syntezuje się cały łańcuch peptydowy krok po kroku od C-końca. W tym celu N-chroniony aminokwas reaguje z reaktywną grupą polimeru. Ze związanego kowalencyjnie z polimerem aminokwasu usuwa się osłonę grupy α-aminowej i otrzymany aminoacylopolimer przereagowuje z następnym N-chronionym aminokwasem. W zasadzie łańcuch peptydowy przedłużany jest krok po kroku we wnętrzu matrycy polimeru. Produkt reakcji związany jest w sposób trwały z nośnikiem, a nadmiar odczynników oraz produkty uboczne reakcji usuwane są za pomocą zwykłego przemywania i sączenia.
W ostatnim etapie syntezy rozszczepiane jest wiązanie kowalencyjne między C-końcowym aminokwasem zsyntezowanego łańcucha peptydowego, a grupą na nośniku, z którą był związany. Nierozpuszczalny nośnik można oddzielić od znajdującego się w roztworze polipeptydu przez zwykłe odsączenie. Prostota operacji technicznych (zastąpienie pracochłonnych etapów wytrącania i oczyszczania, niezbędnych w konwencjonalnej syntezie, zwykłym sączeniem) oraz możliwość automatyzacji procesu stanowią bezdyskusyjne zalety tej metody. Znaczną wadę w początkowym etapie rozwoju syntezy na nośniku stanowił problem otrzymywania czystych peptydów. Wynikało to z braku ilościowego przebiegu reakcji przyłączania i odblokowywania aminokwasów na poszczególnych etapach syntezy. Otrzymywane na nośniku stałym produkty końcowe wymagały żmudnego procesu oczyszczania.
Literatura:
Jakubke HD, Jeschkeit H: ”Aminokwasy, peptydy białka” (1989) wydanie drugie PWN Warszawa
2. Atherton E, Sheppard RC: “Solid Phase Peptide Synthesis: A Practical Approach” (1989) IRL Press Oxford, England
3. Fields GB, Noble RL “Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids” (1990) Int. J. Peptide. Protein. Res. 35: 161- 214
4. Jones J: “The Chemical Synthesis od Peptides” (1994) Clarendon Press, Oxford, England 5. Chan WC, White PD: “Fmoc Solid Phase Peptide Synthesis: A Practical Approach” (2000) Oxford University Press, Oxford, England
6. Jones J: “Amino Acid and Peptide Synthesis” (2002) Oxford University Press, Oxford, England
7. Shawn Doonan: ”Białka i peptydy” (2008), PWN Warszawa
Skróty użyte w tekście
AA – aminokwas
AcOH – kwas octowy
Boc – t-butyloksykarbonyl
DCCI – N,N’-dicykloheksylokarbodiimid
DCM – dichlorometan
DIC − N,N’-diizopropylokarbodiimid
DIPEA – diizopropyloetyloamina
DMF – N,N-dimetyloformamid
Et2O – eter dietylowy
EtOH − etanol
Fmoc − 9-fluorenylometoksykarbonyl
HOBt − 1-hydroksybenoztriazol
HPLC – wysokosprawna chromatografia cieczowa
MALDI-MS − spektrometria mas z jonizacją przez desorpcję w matrycy
MeOH – metanol
n-BuOH − n-butanol
NMP − N-metylo-2-pirolidon
tBu − t-butyl
TEA – trietyloamina
TFA – kwas trifluoroocotowy
TIPS − triizopropylosilan
Synteza peptydów na nośniku stałym metodą Fmoc/tBu
Synteza peptydów na nośniku stałym (żywicy) metodą Fmoc/tBu składa się z kilku etapów. Pierwszy etap syntezy stanowi proces przyłączenia C-końcowego aminokwasu do „linkera” żywicy. Kolejny etap stanowi proces wydłużanie łańcucha polipeptydowego polegający na cyklicznym przyłączaniu kolejnych reszt aminokwasowych. Ostatni etap syntezy polega na odszczepiania peptydu od żywicy z jednoczesnym usunięciem grup ochronnych z łańcuchów bocznych aminokwasów. Przykładowo jeden cykl wprowadzania reszty aminokwasowej podczas wydłużaniu łańcucha peptydowego składa się z następujących etapów:
A. Przemywanie:
2×4 mL DMF, 0,5 min.
B. Dwuetapowe usunięcie osłony Fmoc:
1×3 mL 20% piperydyna/DMF z dodatkiem 1% tritonu X-100, 5 min
1×5 mL 20% piperydyna/DMF z dodatkiem 1% tritonu X-100, 15 min
C. Przemywanie:
2×4 mL DCM, 0,5 min
2×4 mL DMF, 0,5 min
2×4 mL DCM, 0,5 min
3×4 mL DMF, 1 min
3×4 mL DCM, 1 min
D. Potwierdzenie obecności wolnych grup aminowych jednym z testów:
Kaisera,
chloranilowym
lub fluorescaminowym 3
E. Acylowanie:
Pierwsza reakcja przyłączania Fmoc-AA prowadzona jest 90 min, z użyciem trzykrotnego molowego nadmiaru chronionego aminokwasu względem osadzenia żywicy.
Drugie acylowanie (w przypadku pozytywnego testu na obecność wolnych grup aminowych) prowadzona jest 60 min, z użyciem dwukrotnego molowego nadmiaru chronionego aminokwasu.
We wszystkich przypadkach acylowanie prowadzone jest w mieszaninie rozpuszczalników DMF:NMP 1:1 (v/v) z 1% dodatkiem Tritonu X-100.
F. Przemywanie:
3×4 mL DMF, 1 min
3×4 mL DCM, 1 min
G. Monitorowanie reakcji acylowania:
Test na obecność wolnych grup aminowych (Kaisera, chloranilowy lub fluorescaminowy). Pozytywny wynik testu powoduje powtórne acylowanie począwszy od punktu E. Wynik negatywny kończy proces przyłączania chronionego aminokwasu.
W ostatnim etapie syntezy peptydów usuwana jest osłona Fmoc z grupy α-aminowej N-końcowego aminokwasu według procedury przedstawionej w punktach A-C. Na końcu żywica przemywa jest niewielką ilością EtOH oraz Et2O i suszona się w eksykatorze próżniowym nad P2O5 i NaOH do stałej masy.
Odszczepianie peptydów od żywicy prowadzone jest zwykle z jednoczesnym usunięciem osłon z grup funkcyjnych łańcuchów bocznych aminokwasów, np. z zastosowaniem mieszaniny TFA/Fenol/H2O/TIPS (88:5:5:2). Reakcja, z zastosowaniem powyższej mieszaniny, prowadzona jest przez 120 min w temperaturze pokojowej w atmosferze gazu obojętnego (argonu). Po tym czasie żywica jest odsączana i przemywana dwukrotnie niewielkimi ilościami TFA, a następnie z przesączu wytrącany jest peptyd za pomocą zimnego Et2O. Wytrącony peptydy jest odsączany i po rozpuszczeniu w wodzie lub 20% AcOH liofilizowany.
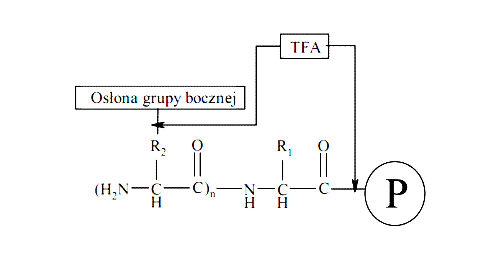
Ryc. 1. Schemat procesu odszczepienia peptydu od żywicy za pomocą TFA
Poszczególne etapy syntezy można przedstawić schematycznie w następujący sposób:
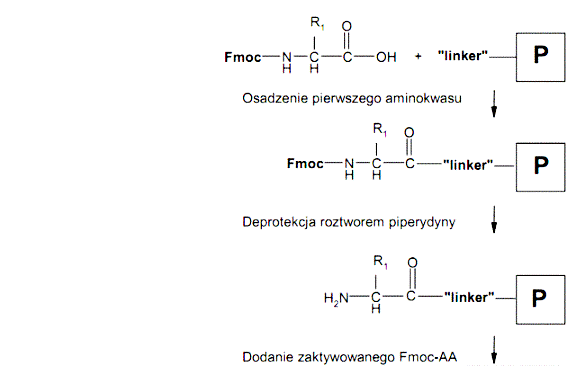
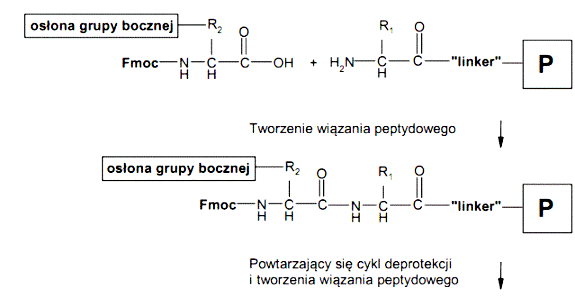
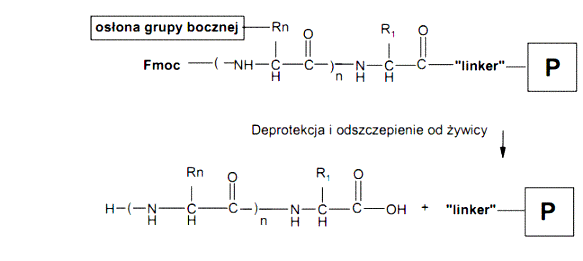
Ogólny schemat syntezy peptydów na nośniku stałym metodą Fmoc/tBu
Synteza peptydów na nośniku stały
Odczynniki do syntezy peptydów:
1. Żywica:
Chloro-(2’-chloro)trityl polystyrene resin (o osadzeniu grup funkcyjnych 1 mM/g)
2. Fmoc-AA:
Fmoc-Gly [M.cz. = 297,3]
Fmoc-Phe [M.cz. = 387,4]
Fmoc-Gln [M.cz. = 368,4]
Fmoc-Trp [M.cz. = 426,5]
Fmoc-Leu [M.cz. = 353,4]
3. Czynniki aktywujące:
HOBt [M.cz. = 135]
DIC [M.cz. = 126; d = 0,806 g/L; 1 mM = 156 µL]
4. Rozpuszczalniki:
DIPEA [1 mM = 174 µL]
MeOH
DMF
DCM
EtOH
Piperydyna
EtO2
AcOH
n-heksan
5. Odczynniki do testu Kaisera
n-BuOH − 2 mL
ninhydryna – 50 mg
pirydyna – 5 mL
KCN – 100 µL 0,001 M wodnego roztworu
fenol − 4 g
Sprzęt laboratoryjny:
1. Zestaw do wysokosprawnej chromatografii cieczowej składający się z detektora UV, pompy oraz komputera z oprogramowaniem
2. Spektrometr firmy Bruker BIFLEX III MALDI-TOF MS
3. Waga analityczna
4. Wytrząsarka
5. Wyparka rotacyjna próżniowa
6. Płaszcz grzejny zaopatrzony w stalowy kołnierz i termometr do testu Keisera
7. Kolby ssawkowe stożkowe 6
8. Kolby okrągłodenne
9. Pipety Pasteura
10. Pipety wielomiarowe
11. Cylindry miarowe
12. Lejki
13. Zlewki
14. Kolby stożkowe płaskodenne
15. Ampułki
16. Probówki Eppendorfa
17. Sączki
18. Kolumienki do ekstrakcji w fazie stałej
19. Kapilary
20. Łopatki dentystyczne
21. Igły lekarskie
22. Balony gumowe
23. Dreny teflonowe oraz typu PEEK
24. Korki teflonowe zaopatrzone w zawory

Zdjęcie 1. Zestaw do prowadzenia syntezy peptydów na nośniku stałym
Synteza peptydu o określonej sekwencji
1. Czynności wstępne – przygotowanie rozpuszczalników i odczynników
W pracy z żywicą, aż do momentu przyłączenia pierwszego Fmoc-AA należy zachować warunki bezwodne, bez śladów alkoholi (np. EtOH, MeOH). Związki te powodują wymianę atomów chloru na żywicy na grupę hydroksylową, wtedy nie możliwe jest przeprowadzenie reakcji w opisany sposób. Dlatego należy bezwzględnie pracować w suchych rękawicach, stosować suche szkło i wszystkie używane do syntezy narzędzia typu łopatki, końcówki do pipet.
Rozpuszczalniki co najmniej na 24 godziny przed użyciem odpowiednio przygotować:
DCM zasypać bezwodnym MgSO4 i K2CO3, wsypując do butelki warstwę około 4 cm soli. Przed użyciem niewielkie ilości odsączyć od środków suszących. DMF i DIPEA zasypać sitami molekularnymi typu A4, wyprażonymi w temperaturze 150oC przez 3 godziny i ostudzonymi w eksykatorze próżniowym. Stosować bezpośrednio znad sit, ostrożnie pobierając z butelki pipetą tak aby nie mieszać sit z odczynnikiem. Odczynniki do testu Kaisera należy przygotować w trzech oddzielnych buteleczkach (każda o pojemności około nie większej niż 10 mL) zaopatrzonych w wkraplacz.
Poszczególne buteleczki powinny zawierać:
1. 1 mL roztworu ninhydryny (50 mg ninhydryny na 1 mL n-BuOH)
2. 1 mL roztworu fenolu (4 g fenolu na 1 mL n-BuOH)
3. 1 mL roztworu KCN (100 µL 0,01 M KCN w wodzie na 5 mL pirydyny)
2. Osadzenie pierwszego C-końcowego aminokwasu
Odważyć 2 eq. (0,3 mM) Fmoc-AA do małej kolby płaskodennej o pojemności 10-20 mL i rozpuścić w minimalnej objętości (około 2-3 mL) świeżo odsączonego znad bezw. MgSO4 i K2CO3 DCM*, do roztworu dodać kroplami DMF, aż do całkowitego rozpuszczenia Fmoc-AA. Do roztworu dodać 8 eq. (1,2 mM) DIPEA (Uwaga! 1 mM DIPEA = 174 µL), a następnie wprowadzić do strzykawki zawierającej 150 mg żywicy wstępnie przemytej DCM (2×4 mL DCM, 2 min). Reakcje prowadzić 1 godzinę, mieszając zawartość strzykawki azotem (balon, ze strzykawką napełniony azotem z końcówką z drenu PEEK). Po 1 godzinie do mieszaniny dodać 2 mL MeOH i reakcję prowadzić jeszcze 15 min. Po tym czasie zawartość strzykawki odsączyć i przemyć zgodnie z poniższym opisem:
3×4 mL DCM : MeOH : DIPEA (17 : 2 : 1; v/v/v), 2 min
3×4 mL MeOH, 2 min
2×4 mL DCM, 2 min
2×4 mL DMF, 1 min
2×4 mL DCM, 1 min
* ze względu na małą rozpuszczalność Fmoc-Gln w DCM należy zastosować wyłącznie DMF, zamiast DCM 8
3. Przyłączanie kolejnych reszt aminokwasowych Fmoc-AA:
a) wstępne przemywanie żywicy (jeżeli żywica była przechowywane w lodówce do kolejnych zajęć)
2×4 mL DCM, 1 min
b) usuwanie osłony Fmoc (deprotekcja)
1×4 mL 20% piperydyna/DMF, 5 min
1×4 mL 20% piperydyna/DMF, 10 min
c) przemywanie żywicy po deprotekcji
3×4 mL DMF, 2 min
3×4 mL DCM, 2 min
2×4 mL MeOH, 2 min
2×4 mL DCM, 1 min
d) test na obecność wolnych grup aminowych (test ninhydrynowy – Keisera):
roztwór 1. 5% ninhydryna w n-BuOH (v/v)
roztwór 2. 80% fenol w n-BuOH (v/v)
roztwór 3. KCN w pirydynie (np. 2 mL 0,001 M roztworu KCN w 98 mL pirydyny)
Niewielką ilość żywicy umieścić w szklanej ampułce (wykonanej z bezbarwnego szkła), dodać po 2 krople każdego roztworu i ogrzewać 5 min w temperaturze 100-105°C. Barwa niebieska ziaren żywicy bądź roztworu wskazuje na obecność wolnych grup aminowych (wynik testu pozytywny).
e) aktywacja i przyłączanie kolejnego Fmoc-AA
Odważyć 4 eq. (0,6 mM) Fmoc-AA do małej kolby płaskodennej o pojemności 10-20 mL i rozpuścić w minimalnej objętości (około 2-3 mL) świeżo odsączonego znad bezwodnego MgSO4 i K2CO3 DCM*, do roztworu dodać kroplami DMF, aż do całkowitego rozpuszczenia Fmoc-AA. Do roztworu dodać 4 eq. (0,6 mM) HOBt, a następnie 4 eq. (0,6 mM) DIC (Uwaga! 1 mM DIC = 156 µL).
Mieszaninę wprowadzić do strzykawki zawierającej żywicę wstępnie przemytą DCM. Reakcje prowadzić 1 godzinę, mieszając zawartość strzykawki azotem (balon, ze strzykawką napełniony azotem z końcówką z drenu PEEK). Po tym czasie zawartość strzykawki odsączyć i przemyć zgodnie z opisem poniżej.
f) przemywanie żywicy po aktywacji i przyłączeniu Fmoc-AA
3×4 mL DMF, 2 min
3×4 mL DCM, 2 min
2×4 mL MeOH, 2 min
2×4 mL DCM, 1 min
* ze względu na małą rozpuszczalność Fmoc-Gln w DCM należy zastosować wyłącznie DMF, zamiast DCM 9
g) test na obecność wolnych grup aminowych (test ninhydrynowy – Keisera)
Barwa niebieska (szara) ziaren żywicy bądź roztworu wskazuje na obecność wolnych grup aminowych. Oznacza to, że proces przyłączania Fmoc-AA nie przebiegł całkowicie i należy go powtórzyć zgodnie z procedurą opisaną w punkcie 3e), a następnie przemyć żywicę zgodnie z procedurą opisaną w punkcie 3f).
Negatywny wynik testu ninhydrynowego oznacza, że można przejść do etapu przyłączenia kolejnej reszty aminokwasowej, zgodnie z procedurą opisaną w punkcie 3.
4. Końcowa deprotekcja − usunięcie osłony Fmoc z N-końcowej reszty AA w peptydzie zgodnie z procedurą opisaną w punktach 3a−d
Po usunięciu osłony Fmoc z N-końcowego aminokwasu w peptydzie wskazane jest żywicę wysuszyć w eksykatorze próżniowym nad KOH i określić przyrost masy żywicy po zakończeniu syntezy.
5. Odszczepienie peptydu od żywicy
Strzykawkę zawierającą peptydylożywicę wraz z korkiem teflonowym umieścić w nasadce do sączenia pod zmniejszonym ciśnieniem nałożonej na zważoną i czystą kolbę okrągłodenną o pojemności 100 mL. Do strzykawki wprowadzić 4 mL 50% AcOH w DCM. Reakcje prowadzić 1 godzinę, mieszając zawartość strzykawki azotem (balon, ze strzykawką napełniony azotem z końcówką z drenu PEEK). Następnie zawartość strzykawki odsączyć i przemyć 3×1 mL 50% AcOH w DCM. Z przesączu odparować DCM i AcOH z dodatkiem n-heksanu, do zaniku zapachu kwasu. Do kolby dodać wodę a następnie zawartość kolby zamrozić w mieszaninie suchy lód/aceton i zliofilizować. Po liofilizacji zważyć i określić masę otrzymanego surowego peptydu.
Analiza tripeptydu
1. Analiza HPLC (ocena czystości otrzymanego peptydu)
Niewielką ilość peptydu przenieść do małej probówki Ependorfa i rozpuścić w niewielkiej objętości MeOH.
Warunki analizy:
– chromatograf cieczowy składający się z pompy Knauer K-1001 i detektora UV Knauer K-2001;
– kolumna: Vydac C18 (5 µm), o wymiarach 250×4,6 mm;
– objętość dozowania: 10 µl;
– temperatura kolumny: 25°C;
– faza ruchoma: ACN : H2O (10 : 90 v/v/) z dodatkiem 0,1% TFA;
– natężenie przepływu fazy ruchomej: 1 mL/min;
– czas analizy: 20 min;
– warunki detekcji: λ = 220 nm; 10
2. Analiza MALDI-MS (identyfikacja tripeptydu)
Jonizacja przez desorpcję laserową w matrycy (MALDI – Matrix Assisted Laser Desorption Ionization) polega na zmieszaniu analizowanej substancji z roztworem małych cząsteczek organicznych, zwanym matrycą, silnie absorbującą promieniowanie przy długości fali lasera (zwykle w zakresie UV). Napromieniowanie tej mieszaniny laserem po uprzednim odparowaniu rozpuszczalnika powoduje nagromadzenie w niej dużej ilości energii i wzbudzenie elektronów w cząsteczkach matrycy. Jony utworzone przez przeniesienie protonu miedzy fotowzbudzoną matrycą a analizowaną substancją ulegają następnie desorpcji. Technika MALDI-MS umożliwia nie tylko dokładne określenie masy cząsteczkowej peptydu lub białka, lecz również wykrywanie mutacji w białkach, identyfikację i lokalizację modyfikacji potranslacyjnych, określenie struktury i sekwencji, a także sprawdzenie czystości peptydów i białek. Niewielką ilość peptydu przenieść do małej probówki Eppendorfa i rozpuścić w MeOH.
Warunki analizy:
Aparat: spektrometr firmy Bruker BIFLEX III MALDI-TOF MS
Jonizacja: promień lasera azotowego o długości fali 337 nm
Matryca: np. kwas α–cyjano-4-hydroksycynamonowy (CCA)