PRZEDRUK, oryginał dostępny pod adresem www
Tytuł oryginalny: Materiały dodatkowe – kwasy i pochodne
Autor: dr Marek Żylewski
Uniwersytet Jagielloński (www)
Wydział Farmaceutyczny Collegium Medicum (www)
Katedra Chemii Organicznej (www)
Kierownik: Prof. UJ, dr hab. Marek Cegła
Adres:
ul. Medyczna 9
30-688 Kraków
Kontakt: tel. 012 620 55 00
Kwasy karboksylowe – kwasowość
Kwasy karboksylowe są kwasami o stosunkowo niewielkiej mocy. Ich stała dysocjacji najczęściej mieści się w zakresie 10-4 – 10-5. Obecność podstawników elektronoakceptorowych może w sposób zdecydowany zwiększyć moc kwasu karboksylowego poprzez stabilizację anionu powstałego w wyniku dysocjacji grupy karboksylowej. Wprowadzenie atomu chloru do cząsteczki kwasu octowego zwiększa jego moc 100-krotnie, natomiast kwas trifluorooctowy należy już do kwasów o dość sporej mocy (pK=0,2), będąc kwasem silniejszym od kwasu fosforowego(V) ale słabszym od kwasu azotowego(V). Podobnie na moc kwasu aromatycznego wpływają podstawniki obecne w pierścieniu, przy czym należy pamiętać, że ich wpływ jest największy, jeśli występują one w pozycji orto lub para względem grupy karboksylowej.
Reakcje redox kwasów karboksylowych
Proste kwasy karboksylowe są zupełnie odporne na działanie nawet silnych środków utleniających i dlatego można je stosować do ich otrzymywania w procesie utleniania alkoholi czy aldehydów. Istnieją jednak kwasy bardzo podatne na utlenianie. Najłatwiej utlenianiu ulega kwas mrówkowy, który, podobnie jak aldehydy, ulega reakcji lustra srebrnego, redukując odczynnik Tollensa. Sam przy tym utlenia się do CO2. Kolejnym przykładem kwasu ulegającego łatwo utlenieniu jest kwas szczawiowy, który pod wpływem KMnO4 utlenia się do CO2. Reakcja ta jest wykorzystywana do ustalania stężenia KMnO4 w roztworze na potrzeby manganometrii. Szczególnym przypadkiem utleniania jest reakcja kwasów z nadtlenkiem wodoru. Prowadzi ona do otrzymania peroksokwasów (nadkwasów) służących jako środki utleniające w licznych syntezach organicznych:
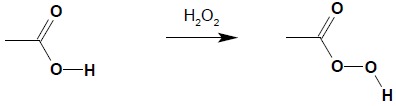
Reakcja ta jest reakcją substytucji nukleofilowej przy karbonylowym atomie węgla, w której wymianie ulega grupa -OH na grupę -OOH
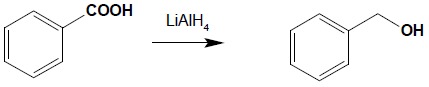
Grupa karboksylowa bardzo trudno ulega redukcji, tylko pod wpływem najsilniej działających reduktorów. Kwasy udaje się zredukować do alkoholi jedynie za pomocą LiAlH4 lub B2H6 – bez udziału tych odczynników bezpośrednia redukcja grupy karboksylowej nie jest możliwa:
Reakcje dekarboksylacji
Usunięcie grupy karboksylowej na drodze reakcji dekarboksylacji prowadzi do otrzymania, w najprostszych warunkach, produktu, w którym w miejscu grupy COOH pojawia się atom wodoru. Powodzenie tej reakcji w sposób ścisły zależy od budowy wyjściowego kwasu.
Prosta dekarboksylacja niepodstawionych kwasów tłuszczowych (alifatycznych) nie jest możliwa do przeprowadzenia, ponieważ w jej trakcie powstają złożone mieszaniny węglowodorów. Wyjątkiem jest kwas octowy, którego sól sodowa reagując z mieszaniną NaOH i CaO daje metan:
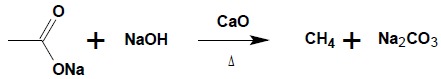
Reakcja ta służy jako dogodna metoda otrzymywania czystego metanu. Znacznie łatwiej ulegają dekarboksylacji kwasy aromatyczne, jednak ta reakcja przeważnie nie znajduje praktycznego zastosowania (odpowiednie związki aromatyczne są łatwiej dostępne niż wyjściowe kwasy).
Największe znaczenie praktyczne ma dekarboksylacja β-oksokwasów i kwasów zawierających dwie grupy karboksylowe przy jednym atomie węgla (pochodne kwasu malonowego). W pierwszym przypadku produktami są ketony:
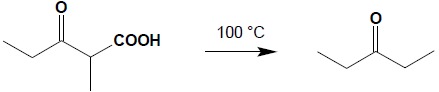
Wyjściowe β-oksokwasy można łatwo otrzymać na drodze kondensacji Claisena i hydrolizie powstałego estru. Stanowi to razem kolejną, dogodną metodę otrzymywania ketonów, często o urozmaiconej budowie.
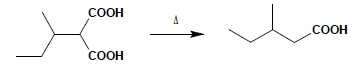
W wyniku dekarboksylacji pochodnych kwasu malonowego otrzymuje się kwasy monokarboksylowe:
Estryfikacja kwasów karboksylowych
Reakcja estryfikacji jest typową reakcją odwracalną, w której stała równowagi często jest rzędu jedności. Reakcja ta wymaga katalizy kwaśnej i rozpoczyna się od protonowania karbonylowego atomu tlenu:
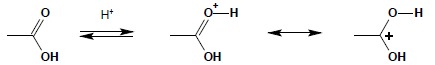
Dalej następuje atak nukleofilowy pary elektronowej tlenu grupy OH alkoholu na karbonylowy atom węgla:
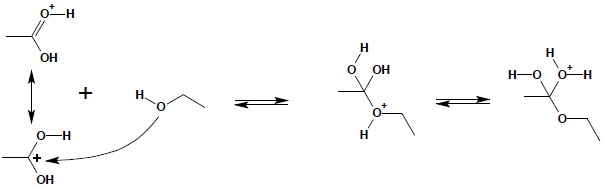
W dalszym ciągu reakcji odłączeniu ulega cząsteczka wody oraz proton:
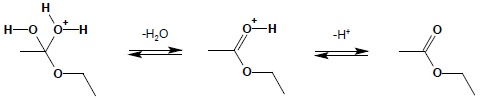
Ze względu na odwracalność każdego z przedstawionych powyżej etapów mechanizm syntezy estrów jest jednocześnie mechanizmem ich hydrolizy w środowisku kwaśnym. Otrzymywanie estrów w wyniku bezpośredniej syntezy jest często wykorzystywane w praktyce, przy czym najlepsze rezultaty (ze względu na położenie stanu równowagi) otrzymuje się, jeżeli w trakcie prowadzenia procesu można jednocześnie usuwać produkt reakcji np., na drodze destylacji.
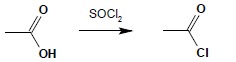
Otrzymywanie chlorków kwasowych
Działając na kwas karboksylowy SOCl2, PCl3 lub PCl5 otrzymuje się jego reaktywną pochodną – chlorek kwasowy, szeroko stosowany środek acylujący:
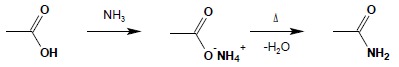
Reakcja z amoniakiem
Działając na kwas karboksylowy amoniakiem, otrzymuje się sól amonową tego kwasu. Sole te na drodze ogrzewania można przekształcić w amidy kwasowe:
Fluorowcowanie w pozycji α
Reakcja wprowadzania atomu chloru lub atomu bromu w pozycję α zachodzi łatwo z udziałem czerwonego fosforu jako katalizatora:
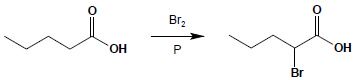
Reakcja ta jest bardzo ważna z punktu widzenia syntezy organicznej, ponieważ otwiera ona możliwość zastosowania tak otrzymanej halogenopochodnej kwasu do przekształcenia ją w inne pochodne (np. aminokwasy, hydroksykwasy) na drodze substytucji nukleofilowej.
Otrzymywanie kwasów karboksylowych
Uzyskanie grupy karboksylowej jest możliwe na wielu drogach:
a) utlenianie alkoholi I-rzędowych i aldehydów:
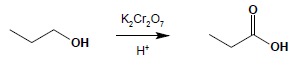
b) utlenianie łańcuchów bocznych węglowodorów aromatycznych
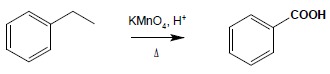
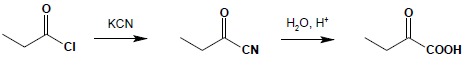
c) hydroliza nitryli:
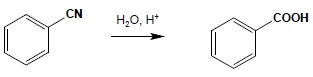
d) reakcja związków Grignarda z CO2:
![]()
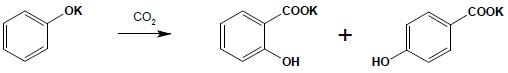
Synteza Kolbego
Synteza ta służy głównie do otrzymywania kwasu salicylowego i dalej z niego – aspiryny, czyli kwasu acetylosalicylowego. Reakcja polega na addycji dwutlenku węgla do soli sodowej fenolu:
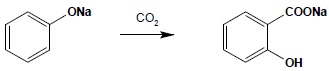
Jest to reakcja o nie do końca poznanym mechanizmie, w której dość istotną rolę gra kation metalu. Użycie w miejsce soli sodowej soli potasowej powoduje otrzymanie mieszaniny dwóch izomerów:
Chlorki i bezwodniki kwasowe
1. Reaktywność pochodnych kwasów karboksylowych
Chlorki i bezwodniki kwasów karboksylowych należą do najaktywniejszych środków acylujących, czyli umożliwiających wprowadzenie grupy acylowej do np. amin (synteza amidów) czy alkoholi (synteza estrów). Aktywność kwasów i ich pochodnych jako środków acylujących można zestawić w poniższy szereg:
chlorki kwasowe > bezwodniki > estry > kwasy karboksylowe.
Ze względu na swą dużą reaktywność w wielu reakcjach chlorki i bezwodniki reagują w ten sam sposób, zatem ich reakcje będą opisywane łącznie.
2. Hydroliza
Chlorki i bezwodniki bardzo łatwo reagują z wodą, dając odpowiednie kwasy. Reakcja ta nie ma żadnego znaczenia preparatywnego (chlorki i bezwodniki otrzymuje się z odpowiednich kwasów, a nie na odwrót), natomiast stanowi istotne ograniczenie w preparatywnym wykorzystaniu tych związków. Aby zapewnić jak najwyższe wydajności rozlicznych procesów, w których substratami są chlorki czy bezwodniki kwasowe należy zapewnić bezwodne środowisko reakcji (poza nielicznymi wyjątkami).
3. Reakcja z alkoholami i fenolami.
W wyniku reakcji chlorków i bezwodników kwasowych z alkoholami i fenolami otrzymuje się estry:
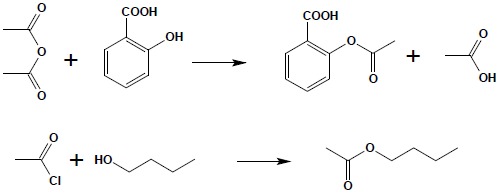
Ze względu na nieodwracalny charakter tej reakcji jest to jedna z najdogodniejszych metod syntezy estrów, będąca jednocześnie metodą z wyboru dla otrzymywania estrów fenoli (fenole nie reagują z kwasami w bezpośredniej syntezie).
4. Reakcja z amoniakiem i aminami.
Chlorki i bezwodniki kwasowe są bardzo dogodnymi substratami do syntezy amidów zarówno niepodstawionych (reakcja z amoniakiem) jak i N-podstawionych (reakcja z aminami I- i II-rzędowymi):
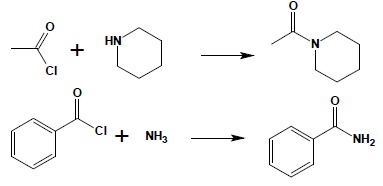
5. Reakcje z innymi odczynnikami nukleofilowymi
Bezwodniki a szczególnie bardziej od nich reaktywne chlorki kwasowe reagują również z wieloma innymi odczynnikami nukleofilowymi, dając różne pochodne. Jednym z ważnych zastosowań jest otrzymywanie nitryli α-oksokwasów jako substratów do otrzymywania odpowiednich kwasów:
6. Reakcja chlorków kwasowych i bezwodników ze związkami Grignarda
Chlorki i bezwodniki kwasowe reagują ze związkami Grignarda w sposób charakterystyczny dla związków karbonylowych. W pierwszym etapie reakcji następuje addycja cząsteczki związku Grignarda do grupy karbonylowej, zgodnie z rozkładem ładunku w obu substratach:
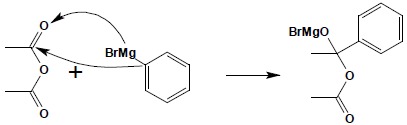
W następnym etapie odłączeniu ulega sól podwójna magnezu (w przypadku bezwodników jest bromek acylowomagnezowy – w omawianym przykładzie bromek octan magnezu; w przypadku chlorków – bromek chlorek magnezu) i powstaje cząsteczka ketonu. Keton natychmiast reaguje z kolejną cząsteczką związku Grignarda, dając alkoholan:
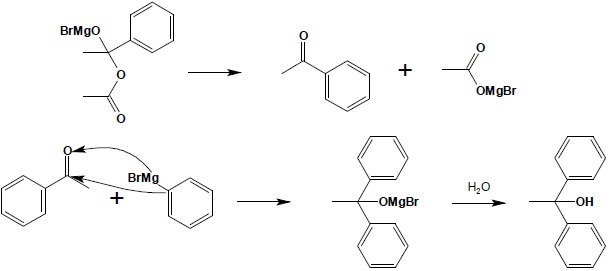
Z powstałego alkoholanu uzyskuje się wolny alkohol na drodze hydrolizy.
7. Acylowanie pierścieni aromatycznych w reakcji Friedela-Craftsa
Chlorki kwasowe (również bezwodniki) reagują z układami aromatycznymi, w obecności AlCl3 jako katalizatora dając ketony:
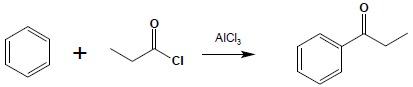
Reakcja podlega tym samym ograniczeniom co reakcja alkilowania pierścieni aromatycznych.
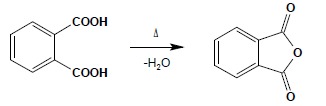
8. Otrzymywanie chlorków i bezwodników kwasowych
Chlorki kwasowe otrzymuje się w wyniku reakcji kwasów karboksylowych z SOCl2, PCl3 lub PCl5 jak zostało to przedstawione powyżej. Bezwodniki otrzymuje się w wyniku dehydratacji kwasów. W przypadku kwasów dikarboksylowych łatwo powstają cykliczne bezwodniki posiadające pierścienie 5- lub 6-członowe – dehydratacja następuje po podgrzaniu:
Ogólną metodą dehydratacji kwasów jest reakcja kwasu z bezwodnikiem octowym:
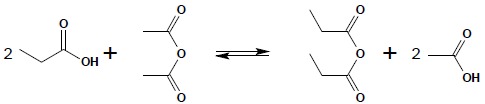
Ponieważ jest to proces równowagowy, o powodzeniu reakcji decyduje możliwość oddestylowania powstającego bezwodnika w trakcie jego powstawania – musi być więc bardziej lotny od kwasu octowego.
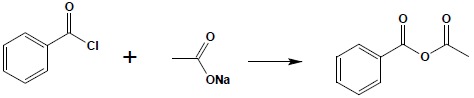
Metodą ogólną syntezy bezwodników zarówno prostych, jak i mieszanych jest reakcja pomiędzy chlorkiem kwasowym a solą kwasu: