
Chromatografia jest techniką analityczną i preparatywną, która umożliwia rozdział mieszaniny na poszczególne składniki lub frakcje. Wykorzystuje ona różnice w zachowaniu się poszczególnych związków w układzie dwufazowym, w którym jedna z faz nie zmienia swojego położenia (faza stacjonarna), druga natomiast porusza się względem pierwszej w określonym kierunku (roztwór rozwijający). W przypadku chromatografii cieczowej (ang. Liquid Chromatography) faza ruchoma jest cieczą.
Układ chromatograficzny
Układ chromatograficzny składa się z trzech składników:
• mieszaniny poddawanej rozdzieleniu;
• fazy stacjonarnej, sorbentu, czyli stałego lub żelowego złoża, które może oddziaływać międzycząsteczkowo ze związkami tworzącymi analizowaną mieszaninę. Istotna dla rozdziału jest zarówno wielkość powierzchni sorpcyjnej, jak i stopień jej obsadzenia grupami funkcyjnymi;
• fazy ruchomej, czyli eluentu lub inaczej czynnika wymywającego. W przypadku chromatografii cieczowej jest nim ciekły rozpuszczalnik, który przenosi analizowaną substancję przez złoże. Dobry eluent powinien zapewniać wystarczającą selektywność układu chromatograficznego, a przy tym nie reagować ani z fazą stacjonarną, ani z substancjami rozdzielanymi (wyłączając zamierzone działania). Powinien on również charakteryzować się możliwie niską lepkością, niskim ciepłem parowania i nie absorbować światła w tych zakresach fali, które będą wykorzystywane przy detekcji.
Proces chromatograficzny to wielokrotna sorpcja i desorpcja substancji z fazy stacjonarnej do fazy ruchomej i odwrotnie, tzn. z fazy ruchomej do stacjonarnej. Różne związki oddziałują z każdą z faz w charakterystyczny dla siebie sposób i z tego względu przemieszczają się z różną szybkością, co umożliwia ich rozdział. Czas ich wędrówki określa się mianem retencji.
Elucja
Proces wymywania składników nazywa się elucją. Istnieją dwa sposoby jej prowadzenia:
• izokratyczna – podczas której skład eluentu jest stały;
• gradientowa – w której dzięki zmianie składu fazy ruchomej następuje wzrost siły elucyjnej (np. skład eluentu w trakcie rozdzielania zmienia się od 10% do 90% metanolu w wodzie). Faza ruchoma o odpowiedniej mocy do rozdzielenia składników o dużej retencji nie rozdziela składników wymywanych na początkowym etapie. Faza ruchoma odpowiednia dla składników o słabej retencji nie wymywa składników silnie zatrzymywanych lub też powoduje ich oddzielenie dopiero po bardzo długim czasie. Elucja gradientowa rozwiązuje ten problem dzięki systematycznemu zwiększaniu siły elucyjnej i stanowi najskuteczniejszy sposób rozwiązania tzw. ogólnego problemu elucji. Stosuje się ją w przypadkach, kiedy analizowana mieszanina składa się z substancji znacznie różniących się od siebie właściwościami sorpcyjnymi.
Wyniki analizy
Wyniki przeprowadzonej analizy przedstawia się zazwyczaj w postaci chromatogramu, czyli wykresu pików odpowiadających poszczególnym składnikom. Obraz ten uzyskuje się dzięki zastosowaniu odpowiedniego detektora.
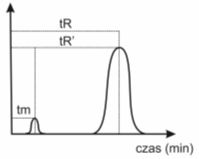
Rys. 1. Chromatogarm elucyjny; tm – zerowy czas retencji lub inaczej czas retencji niezatrzymywanej, wyznaczany dzięki przepuszczeniu przez kolumnę substancji, która nie oddziałuje z fazą stacjonarną; tR -zredukowany czas retencji, czyli okres, jaki upływa między wprowadzeniem próbki, a pojawieniem się maksimum piku rozpatrywanego składnika; tR’ – zredukowany czas retencji, związany z przebywaniem substancji w kolumnie tylko w wyniku oddziaływania tej substancji z fazą stacjonarną (tR’ = tR – tm)
Chromatogram może dostarczać dwojakich informacji:
• jakościowych – liczba pików może oznaczać ilość substancji zawartych w badanej mieszaninie, natomiast ich lokalizacja na chromatogramie pozwala określić rodzaj, tzn. właściwości fizykochemiczne rozdzielanych substancji;
• ilościowych – wielkość rejestrowanego sygnału detektora, tzn. wysokość albo powierzchnia piku chromatograficznego jest funkcją stężenia lub też masy analitu w badanej próbce.
W przypadku chromatografii bibułowej obraz analizy chromatograficznej uzyskuje się bezpośrednio na fazie stacjonarnej, w postaci barwnych palm pochodzących od poszczególnych składników mieszaniny.
Chromatografia preparatywna i procesowa
Chromatografia cieczowa pozwala na identyfikację badanych związków. Jest ona zatem techniką analityczną o ogromnym zakresie zastosowań.
Stanowi ona również jedną z metod wyodrębniania czystych postaci poszczególnych składników mieszanin złożonych. Ze względu na ilość pozyskiwanych substancji wyróżnia się chromatografię preparatywną (dostarczającą niewielkich ilości substancji i jedynie sporadycznie) oraz chromatografię procesową lub inaczej produkcyjną (gdy ilość pozyskiwanego produktu jest znacznie większa, a proces prowadzony jest w sposób cykliczny lub ciągły). Obie znalazły zastosowanie m.in. w izolacji produktów biotechnologii (białek, polisacharydów, fosfolipidów, określonych fragmentów DNA lub RNA), produktów naturalnych pochodzenia roślinnego lub zwierzęcego, lantanowców i transuranowców, izomerów optycznych, produktów syntezy leków, składników kosmetyków, polimerów o niskim stopniu polidyspersyjności, a także użytkowych ilości substancji używanych do badań i mikrosyntez w zakresie chemii organicznej, biochemii, mikrobiologii.
W odróżnieniu od warunków analitycznych, w warunkach chromatografii preparatywnej dąży się do maksymalnego wykorzystania tzw. przeładowania kolumny, dzięki któremu proces rozdzielczy jest bardziej efektywny ekonomicznie.
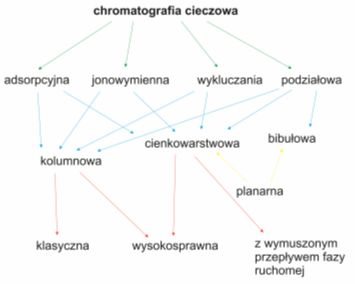
Rys. 2. Rodzaje chromatografii cieczowej
Klasyfikacja chromatografii cieczowej ze względu na różne mechanizmy rozdziału
• Układy chromatograficzne adsorpcyjne (ciecz – ciało stałe), w których wyróżnia się:
Układ faz normalnych (NP), wykorzystujący interakcje pomiędzy polarnymi grupami funkcyjnymi rozdzielanych składników, a polarnymi centrami aktywnymi znajdującymi się na powierzchni fazy stacjonarnej. Podstawowym zjawiskiem decydującym o rozdzielaniu jest adsorpcja na powierzchni bardziej polarnego sorbentu.
Od pierwszych lat stosowania chromatografii cieczowej, fazą stacjonarną były substancje nieorganiczne, takie jak krzemian magnezu, węglan magnezu, tlenek magnezu lub też węglan wapnia i inne. Fazę ruchomą stanowiły natomiast rozpuszczalniki organiczne. Rozwój wysokosprawnej chromatografii cieczowej (HPLC) doprowadził do stosowania organicznych faz stacjonarnych, których właściwości są bardzo zbliżone do adsorbentów nieorganicznych.
Chromatografię w układzie faz normalnych stosuje się jako alternatywę dla układów faz odwróconych podczas rozdzielania związków niejonowych i raczej nisko oraz średnio polarnych, które nie mogą ulec rozpuszczeniu w środowisku wodnym. Dodatkowo, układy faz normalnych posiadają zdolność do wysoce selektywnego rozdzielania izomerów strukturalnych, a w przypadku stosowania chiralnych faz stacjonarnych, również do rozdzielania izomerów optycznych.
Mimo tych zalet, w chromatografii cieczowej dominują obecnie tzw. układy faz odwróconych (RP), czyli takie, w których faza stacjonarna jest mniej polarna niż faza ruchoma. Nazwa ich wynika jedynie z historii chromatografii. Fazę ruchomą stanowi tu mieszanina wody i rozpuszczalnika organicznego. Retencja związku zależy od chemicznej natury cząsteczek substancji, eluentu i sorbentu. Substancje opuszczają kolumnę w kolejności od najbardziej polarnych do niepolarnych, a w szeregach homologicznych od nisko- do wysokocząsteczkowych. Miarą różnic retencji poszczególnych substancji jest ich selektywność hydrofobowa, chemiczna i steryczna.
• Podziałowe (ciecz-ciecz)
Analizowane substancje rozdzielane są pomiędzy dwie ciekłe, niemieszające się fazy: ruchomą i stacjonarną, osadzoną statycznie na nośniku (związaną z nim siłami przylegania), bądź też generowaną w sposób dynamiczny w czasie przepływu eluentu przez kolumnę.
Zgodnie z prawem podziału Nernsta, stosunek stężeń w obu fazach w stanie równowagi jest wielkością stałą i charakterystyczną w danych warunkach dla danej substancji. Oznacza to, że jeżeli stężenie badanego składnika w fazie ruchomej jest zbyt duże, substancja ta przechodzi do fazy ciekłej stacjonarnej i odwrotnie, gdy faza ruchoma ma mniejsze stężenie, składnik częściowo przechodzi z fazy stacjonarnej do fazy ruchomej. Proces ten trwa aż do momentu ustalenia się odpowiedniego stosunku stężeń w obu fazach.
• Chromatografię żelową, wykluczenia sterycznego, sitową
Ten typ chromatografii eliminuje, o ile to możliwe, zjawisko sorpcji. Składniki mieszaniny rozdzielane są na tzw. sitach molekularnych, czyli ziarnach tworzących pory międzyziarnowe przez które płynie ciecz. Zawierają one również mikropory wewnętrzne, w których przestrzeni eluent praktycznie nie płynie. W ten sposób związki rozdzielane są ze względu na swoją wielkość i masę molową.
• Chromatografię jonowymienną
Ten typ chromatografii korzysta z różnic w ładunku wypadkowym odmiennych cząstek, a rozdział mieszaniny następuje w wyniku odwracalnej wymiany jonów z fazą stacjonarną. Cząsteczki o małej gęstości wypadkowych ładunków dodatnich wypływają z kolumny jako pierwsze, a cząsteczki o większej gęstości ładunków – dopiero w następnej kolejności.
Białka naładowane ujemnie (anionowe) można rozdzielać chromatograficznie na kolumnach wypełnionych dodatnio naładowaną dietyloaminoetylocelulozą (DEAE-celulozą). Natomiast białka naładowane dodatnio można rozdzielać na kolumnie z ujemnie naładowaną karboksymetyocelulozą (CM – celulozą).
Do tej grupy chromatografii zalicza się również tzw. technikę wykluczania jonowego, w której mechanizm rozdzielania wykorzystuje zjawisko powstawania tzw. membrany Donnana.
Alternatywą dla chromatografii jonowymiennej jest chromatografia par jonowych, stosowana głównie w układach faz odwróconych, gdy jonowe, albo bardzo silnie polarne fragmenty cząsteczek substancji rozdzielanych są maskowane odpowiednim organicznym przeciwjonem. Powstający w ten sposób kompleks zachowuje się zatem jak substancja neutralna.
• Chromatografię powinowactwa
Technika ta wykorzystuje duże powinowactwo wielu białek do specyficznych grup chemicznych (np. oddziaływanie enzym – koenzym) albo innego typu oddziaływania zachodzące pomiędzy fazą stacjonarną, a rozdzielanymi składnikami (np. tzw. chromatografia “metalopowinowactwa” – oddziaływania Ni…S).
Można ją stosować bez kolumny, tzn. wykonywać rozdzielenie w naczyniu laboratoryjnym albo bezpośrednio w bioreaktorze.
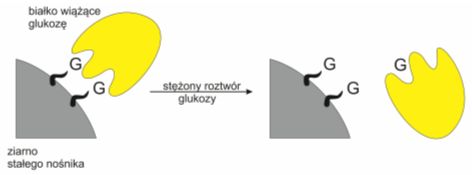
Rys. 3. Chromatografia powinowactwa konkanawaliny A (kolor żółty) na stałym nośniku zawierającym kowalencyjnie przyłączone reszty glukozy. Związane białko można uwolnić dodając stężony roztwór glukozy
Podział chromatografii cieczowej ze względu na sposób prowadzenia rozdziału
Chromatografię cieczową można również podzielić na:
• kolumnową, w której adsorbent stanowi wypełnienie kolumny chromatograficznej.
Składniki mieszaniny przemieszczają się ku dołowi z szybkością zależną od siły oddziaływań ich cząsteczek z adsorbentem. Poszczególne związki wędrują z różną szybkością i dzięki temu można je kolejno odbierać na dole kolumny.
Przepływ fazy ruchomej może być grawitacyjny lub wymuszony za pomocą ciśnienia pod jakim eluent jest wprowadzany do kolumny (ang. High Performance Liquid Chromatography – HPLC, wysokosprawna chromatografia cieczowa).
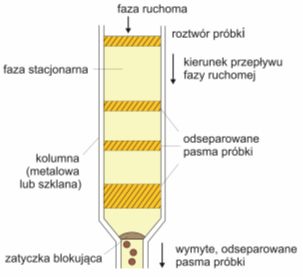
Rys. 4. Kolumna chromatograficzna
• planarną, gdzie faza stacjonarna jest uformowana w postaci płytki lub płasko rozprowadzona na odpowiednim nośniku.
Zalicza się tutaj chromatografię bibułową, w której rozdział substancji wykonuje się na odpowiednio dobranej bibule oraz cienkowarstwową (z ang. Thin Layer Chromatgraphy, TLC), w której sorbent jest osadzony na podłożu, tzn. folii lub płytki wykonanej ze szkła, aluminium lub tworzywa sztucznego.
Chromatografię cienkowarstwową po raz pierwszy wykorzystano w 1938 roku do oznaczania zanieczyszczeń leków. Jej znaczny rozwój nastąpił po 1956 roku, dzięki odkryciu tzw. urządzenia Stahla, umożliwiającego w prosty sposób przygotowanie warstw chromatograficznych na płytkach szklanych. Jego odkrywca był również twórcą podstaw teoretycznych i metodycznych tego typu chromatografii.
Główną zaletą TLC jest możliwość przechowywania płytek z rozdzielonymi substancjami. Pozwala ona również określić stopień rozdzielenia substancji na każdym etapie rozwijania chromatografu, a co za tym idzie przerwać ten proces w dowolnym momencie. W przypadku kolumny chromatograficznej zarówno detekcja składników, jak i ocena stopnia ich rozdzielenia możliwe są dopiero, gdy związki te opuszczą kolumnę.
Znaczącym ulepszeniem okazało się zastosowanie tzw. densytometrów, czyli detektorów służących do automatycznego dozowania próbki oraz komputerowego opracowywania wyników. Możliwości rozdzielcze tej techniki zwiększyło dodatkowo wprowadzenie tzw. wysokosprawnej chromatografii cienkowarstwowej (ang. High Performance Thin Layer Chromatography – HPTLC).
Tab.1. Porównanie chromatografii cienkowarstwowej z wymuszonym przepływem fazy ruchomej (konwencjonalnej) oraz wysokosprawnej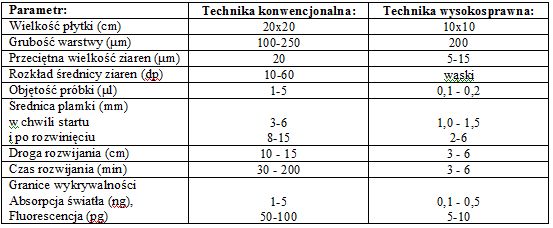
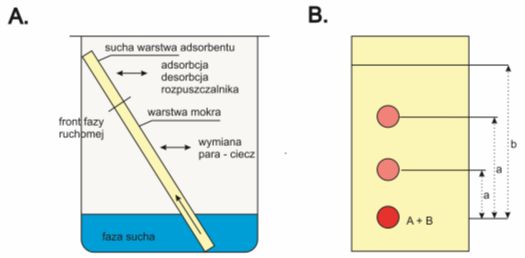
Rys. 5. Schematyczny obraz procesów zachodzących w czasie rozwijania chromatogramu na płytce chromatograficznej w warunkach konwencjonalnej chromatografii cienkowarstwowej (A) oraz schemat chromatografu cienkowarstwowego mieszaniny substancji (B)
Postęp rozwoju chromatografii zarówno cienkowarstwowej jak i kolumnowej wiąże się głównie z zastosowaniem mniejszych ziaren fazy stałej, dostępnością gotowych warstw chromatograficznych oraz instrumentalizacją metody.
Zobacz również:
Chromatografia cienkowarstwowa
Chromatografia jonowymienna
Chromatografia gazowa
Autor: Anna Kurcek
Literatura:
1. Kamiński M., Kartanowicz R., 2004. Chromatografia cieczowa. Politechnika Gdańska.
2. Hierasimczyk K. (tłumaczenie), 2002. Chemia analityczna. Chromatografia. Politechnika Gdańska.
3. 2007. Wysokosprawna chromatografia cieczowa (HPLC). Uniwersytet Gdański.
4. Głoda B. K., 2009. Postępy chromatografii. Wydawnictwo Akademii Podlaskiej.
5. Stryer L., 2003. Biochemia. Wydawnictwo Naukowe PWN. Warszawa.