PRZEDRUK, oryginał dostępny pod adresem www
Tytuł oryginalny: Materiały dodatkowe – alkohole, fenole, etery
Autor: dr Marek Żylewski
Uniwersytet Jagielloński (www)
Wydział Farmaceutyczny Collegium Medicum (www)
Katedra Chemii Organicznej (www)
Kierownik: Prof. UJ, dr hab. Marek Cegła
Adres:
ul. Medyczna 9
30-688 Kraków
Kontakt: tel. 012 620 55 00
Alkohole i fenole
1 Własności kwasowo-zasadowe alkoholi i fenoli
Obecność grupy OH, posiadającej zdolność do dysocjacji, w alkoholach i fenolach decyduje o możliwości pojawienia się właściwości kwasowych i tworzeniu soli. Należy zatem rozpatrzyć moc kwasów, jakimi są alkohole i fenole. Ze względu na fakt, iż związki te należą zasadniczo do słabych kwasów, dobrym odnośnikiem dla ich mocy jest woda. Okazuje się, iż praktycznie wszystkie alkohole są kwasami słabszymi od wody. Do nielicznych wyjątków należą metanol i alkohole o krótkim łańcuchu węglowym, w którym w bezpośrednim sąsiedztwie grupy OH znajdują się podstawniki silnie elektronoakceptorowe (np. atomy F, Cl, czy grupy nitrowe), stabilizujące poprzez wpływ indukcyjny powstały w wyniku dysocjacji anion. Alkohole zatem nie reagują z wodnymi roztworami wodorotlenków i nie można na tej drodze otrzymywać alkoholanów:
ROH + NaOHaq = brak reakcji
Sole alkoholi otrzymuje się poprzez bezpośrednią syntezę alkoholu i odpowiedniego metalu:
ROH + Na = RONa + H2
Ze względu na niewielką kwasowość wyjściowych alkoholi ich sole są bardzo silnymi zasadami (silniejszymi od odpowiednich wodorotlenków) i znajdują przez to szerokie zastosowanie w syntezie organicznej. W wodzie alkoholany ulegają hydrolizie:
RONa + H2O = ROH + NaOH
Fenole z kolei są kwasami silniejszymi od wody, wobec czego można otrzymywać fenolany w reakcji fenolu z odpowiednim wodorotlenkiem.
Ar-OH + NaOH = Ar-ONa + H2O
Właściwości kwasowe fenoli silnie zależą od podstawników znajdujących się w pierścieniu. Obecność podstawników elektronodonorowych obniża kwasowość fenoli, natomiast podstawniki elektronoakceptorowe zwiększają kwasowość. Większa ilość podstawników elektronoakceptorowych może zwiększyć kwasowość fenolu do tego stopnia, iż staje się on kwasem mocniejszym od kwasów karboksylowych. Przykładem jest tzw. kwas pikrynowy – 2,4,6-trinitrofenol będący kwasem silniejszym od prostych kwasów karboksylowych (stała dysocjacji kwasu pikrynowego jest rzędu jedności, podczas gdy kwasu octowego rzędu 10-5). Wpływ podstawników na własności kwasowe fenoli opiera się na efekcie mezomerycznej stabilizacji anionu powstałego w wyniku dysocjacji fenolu. Podstawniki elektronoakceptorowe ułatwiają rozproszenie ładunku ujemnego anionu na atomy węgla pierścienia aromatycznego, zmniejszając gęstość elektronową w pierścieniu aromatycznym.
Podobnie jak woda, alkohole i fenole mogą również pełnić rolę zasady, przyłączając proton w obecności silnych kwasów mineralnych. Powstałe jony noszą nazwę jonów oksoniowych:
ROH + H+ = ROH2+
2 Estryfikacja
Charakterystyczną reakcją alkoholi jest estryfikacja. Reakcja ta podlega katalizie kwasowej i jest procesem równowagowym:
R-COOH + R’-OH = R-COOR’ + H2O
Stała równowagi tej reakcji jest często bliska jedności więc aby uzyskać odpowiednio dużą wydajność procesu, bezpośrednią estryfikację prowadzi się tylko wtedy, jeżeli powstający ester (lub wodę) da się usunąć z mieszaniny reakcyjnej w toku reakcji np. poprzez oddestylowanie. W pozostałych przypadkach otrzymywanie estrów przeprowadza się na innych drogach. Możliwe jest również otrzymywanie estrów kwasów nieorganicznych, jednakże reakcja bezpośredniej estryfikacji jest ograniczona do kwasów siarkowego(VI), azotowego(III), azotowego(V) i ewentualnie kwasu borowego(III).
R-OH + HNO3 = R-O-NO2
W przypadku fenoli reakcji z kwasami siarkowym(VI), azotowym(III) i azotowym(V) nie można przeprowadzić, ponieważ fenole reagują z tymi kwasami wyłącznie w kierunku podstawienia do pierścienia aromatycznego w reakcji substytucji elektrofilowej (nitrowanie czy sulfonowanie).
3 Wymiana grupy OH
W wyniku reakcji z halogenowodorami (HCl, HBr i HI) możliwa jest wymiana grupy hydroksylowej alkoholi na odpowiedni atom fluorowca. Reakcja przebiega najłatwiej z kwasem jodowodorowym, natomiast kwas solny reaguje łatwo jedynie z najbardziej reaktywnymi alkoholami. Reakcja ta jest typową reakcją substytucji nukleofilowej, poprzedzonej protonowaniem grupy OH. Najbardziej reaktywne alkohole III-rzędowe reagują według mechanizmu SN1:
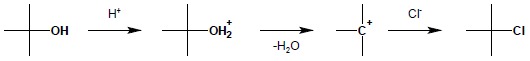
Alkohole I-rzędowe i zazwyczaj II-rzędowe reagują według mechanizmu SN2. Ponieważ HCl reaguje łatwo tylko z najbardziej reaktywnymi alkoholami (IIIrzędowymi, allilowymi i benzylowymi), do wymiany grupy OH na Cl często wykorzystuje się tzw. odczynnik Lucasa. Jest to roztwór ZnCl2 w HCl (ZnCl2 pełni rolę katalizatora w reakcji wymiany). Reakcja ta ma znaczenie również w analizie alkoholi – na podstawie jej wyniku określa się rzędowość badanego alkoholu (alkohole III-rzędowe, allilowe i benzylowe reagują na zimno, natychmiast, II-rzędowe na zimno, lecz dopiero po kilku minutach natomiast I-rzędowe na zimno nie reagują – reakcja zachodzi dopiero na gorąco). Duża reaktywność alkoholi III-rzędowych allilowych i benzylowych wynika z największej trwałości powstającego, jako produkt pośredni, karbokationu, stabilizowanego dodatkowo (w przypadku alkoholi allilowych i benzylowych) poprzez rezonans:
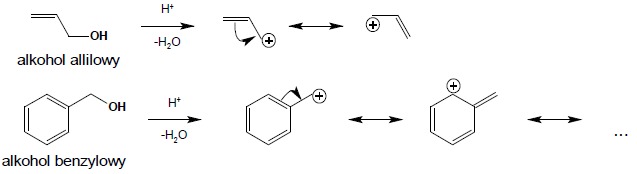
Reakcja wymiany grupy OH na fluorowiec nie jest możliwa w przypadku fenoli.
4 Dehydratacja alkoholi.
W układzie silny kwas mineralny – alkohol występuje szereg procesów równowagowych, pozwalających, w zależności od temperatury, rzędowości alkoholu czy stężenia kwasu, otrzymać rożne produkty. Wyższa temperatura i wyższe stężenia kwasów prowadzi do otrzymania alkenów, w niższych temperaturach produktem głównym mogą być etery:
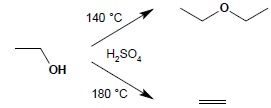
Bardzo wyraźny jest wpływ rzędowości alkoholu na warunki reakcji dehydratacji – im wyżej rzędowy i bardziej rozgałęziony jest alkohol, tym łatwiej następuje dehydratacja do alkenu. I-rzędowy etanol ulega dehydratacji do etenu pod wpływem stężonego H2SO4 w temperaturze 180°C, natomiast III-rzędowy tert-butanol ulega dehydratacji do 2-metylopropenu już w 80°C, pod wpływem 20% kwasu. Na zimno alkohole rozpuszczają się w kwasie siarkowym, dając sole oksoniowe:
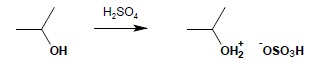
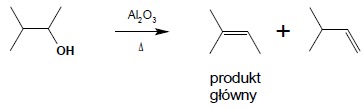
Dehydratacja alkoholi do alkenów przy użyciu kwasu siarkowego, w podwyższonej temperaturze niesie ze sobą niebezpieczeństwo występowania różnych przegrupowań, powstających jako produkt pośredni, karbokationów. Dotyczy to rozgałęzionych alkoholi o bardziej skomplikowanej budowie. Bezpieczniejszą metodą, w której przegrupowań takim można uniknąć jest dehydratacja alkoholi przy użyciu Al2O3 jako katalizatora. Metoda ta wymaga jednak stosowania wysokich temperatur. Pamiętać należy, iż w reakcji dehydratacji prowadzonej zarówno pod wpływem H2SO4, jak i Al2O3 obowiązuje reguła Zajcewa:
5 Przegrupowanie pinakolinowe
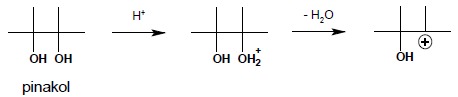
Przegrupowanie to jest przykładem bardzo często spotykanych przegrupowań karbokationów, których siłą napędową jest przemiana mniej trwałego karbokationu w układ bardziej trwały. Przegrupowanie pinakolinowe zachodzi pod wpływem kwasów (np. H2SO4), w podwyższonej temperaturze, dla tzw. wicynalnych dioli, czyli alkoholi posiadających dwie grupy hydroksylowe, obecne przy sąsiednich atomach węgla. Nazwę swą wzięło od nazwy zwyczajowej alkoholu (pinakol), dla którego po raz pierwszy zaobserwowano taką przemianę.
W pierwszym etapie reakcji następuje protonowanie grupy hydroksylowej alkoholu i odłączenie cząsteczki wody z utworzeniem karbokationu:
Następnie następuje przeniesienie grupy węglowodorowej połączonej z sąsiednim atomem węgla, wraz z parą elektronową i jednoczesnym utworzeniem trwalszego od karbokationu jonu oksoniowego:
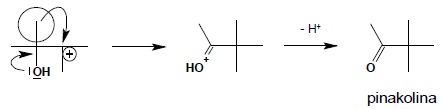
Jon oksoniowy traci następnie proton i powstaje odpowiedni związek karbonylowy. Warunkiem powodzenia reakcji jest powstanie w pierwszym etapie reakcji jak najtrwalszego karbokationu. Ograniczone zatem jest ono do takich tylko dioli, z których w pierwszym etapie reakcji może powstać karbokation III-rzędowy lub lepiej benzylowy czy allilowy. Wynika z tego, iż proste, nierozgałęzione diole typu etano-1,2-diol, butano-2,3-diol itp. nie mogą być użyte jako substrat w tej reakcji – przegrupowanie pinakolinowe dla nich nie zachodzi. Warunek ten pokazuje również kierunek reakcji dla niesymetrycznych dioli – reakcja biegnie zawsze tak aby w pierwszym etapie powstał jak najtrwalszy karbokation. W poniższym przykładzie reakcja biegnie wyłącznie według schematu I, w kierunku tworzenia trwalszego karbokationu benzylowego, stabilizowanego przez rezonans
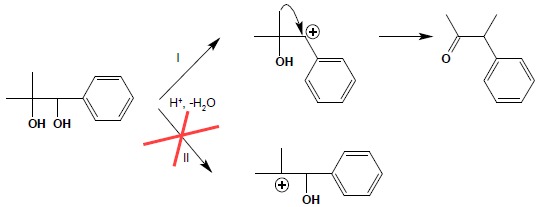
Jeżeli w wyniku odłączenia cząsteczki wody może powstać tylko jeden karbokation (dla układów symetrycznie podstawionych), wtedy o dalszym biegu reakcji decyduje zdolność grup do wędrówki. Obowiązuje reguła (ważna również dla innych przegrupowań karbokationów), iż zdolność wędrówki obrazuje szereg: grupy aromatyczne > rozgałęzione grupy alifatyczne > proste grupy alifatyczne > atomy wodoru. Zastosowanie poniższej reguły obrazuje poniższy schemat reakcji:
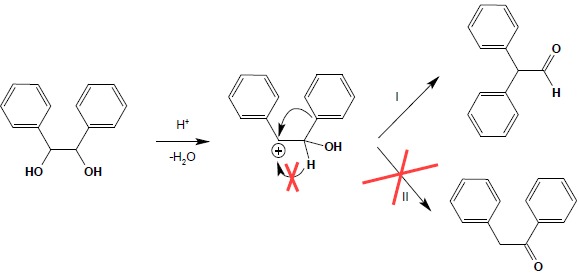
6 Utlenianie alkoholi
Alkohole poddają się łatwo reakcji utleniania za pomocą całej gamy różnych odczynników (manganian(VII) potasu, dwuchromian(VI) potasu, inne związki chromu(VI)) a wynik reakcji zależy od rzędowości alkoholu i użytego utleniacza.
6.1 Alkohole I-rzędowe utleniają się do kwasów karboksylowych lub do aldehydów. Użycie silnego utleniacza (manganian(VII) potasu, dwuchromian(VI) potasu roztwór CrO3 w H2SO4 – odczynnik Jonesa) prowadzi do otrzymania kwasów karboksylowych:
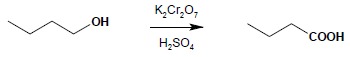
Reakcja biegnie poprzez stadium aldehydu, jednak zatrzymanie reakcji na tym etapie przeważnie nie jest możliwe. Do otrzymania aldehydów służy łagodniej działający utleniacz – PCC (powstaje w wyniku rozpuszczenia CrO3 w mieszaninie HCl z pirydyną w rozpuszczalniku organicznym):
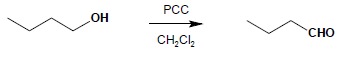
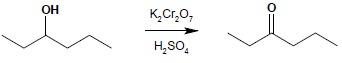
6.2 Alkohole II-rzędowe utleniają się do ketonów:
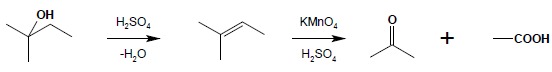
6.3 Alkohole III-rzędowe utlenianiu w zasadzie nie ulegają. Możliwe jest jednak utlenianie takich alkoholi w drastycznych warunkach, biegnące z rozerwaniem łańcucha węglowego. W reakcji tej, zachodzącej w obecności stężonego kwasu siarkowego(VI) następuje wpierw dehydratacja cząsteczki alkoholu, a następnie alken, powstały jako produkt pośredni, ulega w warunkach reakcji utlenieniu z rozerwaniem łańcucha węglowego cząsteczki:
7 Utlenianie fenoli
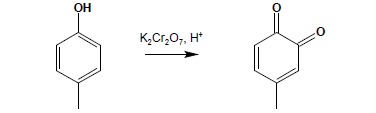
Fenole są związkami ulegającymi utlenieniu stosunkowo łatwo, przy czym podatność układu rośnie wraz ze wzrostem liczby grup hydroksylowych. Mono- i dihydroksylowe fenole utleniają się do chinonów. Utlenianie fenoli monowodorotlenowych wymaga zastosowania bardziej ostrych warunków:
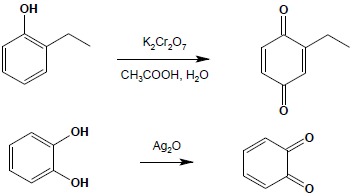
Jeżeli pozycja para fenolu monohydroksylowego jest zajęta, tworzy się pochodna obezochinonu:
Rezorcyna (benzeno-1,3-diol) nie ulega utlenieniu w łagodnych warunkach, ponieważ układ meta-chinoidowy jest niemożliwy do zrealizowania. Rezorcyna ulega utlenieniu w ostrzejszych warunkach ze zniszczeniem układu pierścieniowego do mieszaniny produktów.
Fenole tri- i polihydroksylowe utleniają się już tlenem z powietrza z całkowitym zniszczeniem struktury do mieszaniny produktów takich jak CO, CO2, CH3COOH itd.
8 Otrzymywanie alkoholi i fenoli
8.1 uwodnienie alkenów
8.1.1 addycja wody do wiązania podwójnego w obecności kwasów
8.1.2 hydroksyrtęciowanie (addycja wody do alkenu przy użyciu octanu rtęci jako katalizatora):
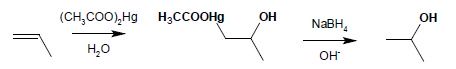
Końcowy efekt reakcji jest identyczny z prostą addycją wody w obecności kwasu (reguła Markownikowa) jednakże reakcja prowadzona tą metodą jest szybsza i bardziej wydajna jako proces laboratoryjny.
8.1.3 hydroborowanie – metoda syntezy alkoholi I-rzędowych z terminalnych alkenów (efekt końcowy to przyłączenie wody do alkenu niezgodnie z regułą Markownikowa). Metoda polega na addycji diboranu do alkenu i utlenieniu powstającego połączenia boroorganicznego:
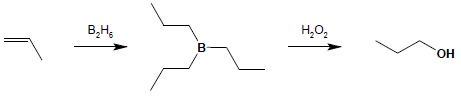
8.2 substytucja nukleofilowa – wymiana fluorowca na grupę OH w halogenopochodnych alifatycznych: R-X + NaOH + H2O → R-OH
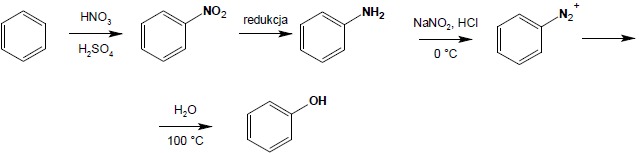
8.3 otrzymywanie fenoli:
8.3.1 stapianie soli arylosulfonowych z NaOH:

8.3.2 reakcja zagotowania soli diazoniowych: