
Autor: Beniamin Grabarek absolwent biotechnologii medycznej na Wydziale Farmaceutycznym z Oddziałem Medycyny Laboratoryjnej Śląskiego Uniwersytetu Medycznego w Katowicach
Komórki nowotworowo zmienione tworzące guz prócz tego, iż posiadają pewien zestaw cech warunkujących im autonomię wzrostu, oddziaływanie z innymi otaczającymi je komórkami tworzącymi mikrośrodowisko guza, charakteryzują się zmienionym metabolizmem [1,2,3]. Pierwsze doniesienia dotyczące odmiennego metabolizmu komórek nowotworowych w stosunku do prawidłowych pojawiły się już na początku XX wieku. Rozwój nowych metod badawczych pozwolił w lepszym stopniu zbadać metabolizm komórek budujących guz, choć ze względu na fakt dużej ilości czynników związanych z pozyskiwaniem energii przez komórki nowotworowe, pojawiły się nowe teorie i doniesienia odnośnie tego tematu. Celem tego artykułu jest przybliżenie charakteru zmian mających wpływ na metabolizm komórek nowotworowych.
W procesie kancerogenezy można wyróżnić cztery podstawowe etapy: preinicjację, inicjację, promocję oraz progresję. Drugi z etapów związany jest z pojawieniem się mutacji, która nie zostaje naprawiona, lecz przekazana dalej kolejnym komórkom [4]. Jej wystąpienie dotyczy najczęściej mitochondrium, prowadząc do zmian w postaci upośledzenia oddychania komórkowego [1].
Komórki nowotworowe cechuje nasilony metabolizm, co jest wynikiem ich nieustannej proliferacji i ma na celu zapewnienie komórkom dostatecznie dużej ilości składników odżywczych i energii do realizacji procesów mających miejsce w trakcie cyklu komórkowego. Pozyskują one energię w wyniku rozpadu wysokoenergetycznych wiązań ATP, którego źródłem jest glikoliza i cykl Krebsa, co znajduje przełożenie we wzmożonym działaniu łańcucha oddechowego [5], prowadząc jako wynik jednoelektornowej redukcji tlenu do powstawania reaktywnych form tlenu (RFT), uszkodzenia mtDNA (mitochondrialny DNA) a w konsekwencji wystąpienia np. wzmożonej proliferacji, apoptozy, nekrozy [1]. Szacuje się, że intensywność procesu glikolizy w komórkach nowotworowych jest ok. 124- krotnie wyższa niż w prawidłowych erytrocytach, dlatego też te komórki wytwarzają ilość energii w procesie oddychania beztlenowego na podobnym poziomie co niezmienione, prawidłowe komórki w procesie komórkowego oddychania tlenowego [5].
W badaniach przeprowadzonych przez Kidd i wsp [6]. oraz John i wsp. [7] podkreślono fakt, iż beztlenowe uzyskiwanie energii jest charakterystyczne dla komórek nowotworowych. Jednym z modeli metabolizmu komórek nowotworowych jest efekt Warburga, wedle którego komórki nowotworowe preferują, nawet w obecności dostatecznie dużych ilości tlenu, oddychanie beztlenowe, wykorzystując jako substrat tylko heksozy (głównie glukozę). Ponadto, jak wskazują dane literaturowe, metabolizm według efektu Warburga, będący prawdopodobnie efektem aktywacji protoonkogenów, zmiany szlaków sygnalizacyjnych jest korzystniejszy dla proliferacji i obserwowany już w szybko proliferujących komórkach prawidłowych.
Inna hipoteza dotycząca metabolizmu nowotworu, określana jako odwrotny efekt Warburga kładzie nacisk na współdziałanie aktywowanych fibroblastów podścieliska guza z komórkami nowotworowymi. Drugim postulatem tej hipotezy jest to, że efekt Warburga dotyczy tylko fibroblastów, w których zachodzące procesy kataboliczne dostarczają komórkom, które uległy procesowi transformacji nowotworowej związków wysokoenergetycznych, takich jak: mleczan, ketony, glutamina, wykorzystywanych przez te komórki do procesów anabolicznych i powstawania ATP w procesie oddychania tlenowego [8].
Wysoka aktywność procesu glikolizy w komórkach nowotworowych wiązana jest z faktem bardzo wydajnego pobierania glukozy przez te komórki. Przekłada się to na nadekspresję przez błonowych transporterów glukozy (GLUT), transportujących tą heksozę niezależnie od energii, zgodnie z gradientem stężeń. Szczególną uwagę poświęca się nadekspresji izoformy 1 GLUT, czyli GLUT1, obserwowaną w wielu typach nowotworów (np. piersi, jelita grubego, żołądka, ślinianek), wskazując na możliwość wykorzystania oznaczania GLUT1 jako wskaźnika progresji zmian nowotworowych, wyłonienia pacjentów, u których zasadne byłoby zastosowanie agresywnych form terapii przeciwnowotworowej [9].
Jak wiadomo, komórki nowotworowe cechuje wysokie tempo proliferacji. Wiąże się to z szybkim przyrostem masy guza oraz niedostatecznym (niepokrywającym zapotrzebowania) dopływem do niego tlenu i substancji odżywczych. W takiej sytuacji dochodzi do aktywacji w komórkach nowotworowych i komórkach ich mikrośrodwowiska czynników transkrypcyjnych aktywowanych niedotlenieniem – HIFs (ang. Hipoxia-inducible factors), w budowie których wyróżnia się podjednostkę α i β. Dodatkowo komórki mikrośrodowiska guza, takie jak: makrofagi, fibroblasty, komórki śródbłonka, komórki układu immunologicznego stanowią dla komórek nowotworowych ochronę przed eliminacją i warunkują im dalszy wzrost. Obecnie znane są 3 izoformy czynnika transkrypcyjnego HIF, które charakteryzują się podobną budową, lecz różnią się choćby miejscem ekspresji. I tak, druga izoforma występuje praktycznie tylko w trzustce i endotelium. Rola i budowa trzeciej izoformy jest nadal badana i mało poznana, wiadomo jednak iż ulega ekspresji w wielu tkankach i zbudowana jest z podjednostek HIF1 β (dimer). Ponadto pierwsza izoforma ulega aktywacji przy stężeniu tlenu poniżej 1% i pełni dominującą rolę w początkowych etapach kancerogenezy, podczas gdy HIF2 indukowany jest stężeniem tlenu z przedziału 2-5%. W warunkach dostatecznej dyfuzji tlenu, jednostka HIF1α podlega procesowi hydroksylacji, przyłaczeniu białka vHL (białko von Hippel – Lindau) i ubikwitynacji, co przekłada się na degradację tej podjednostki jak i całego czynnika HIF1. Jednak gdy poziom tlenu jest na poziomie poniżej 1%, podjednostka α po przejściu z cytoplazmy do jądra komórkowego przyłącza się do podjednostki β tego czynnika, która jest stabilna i tlenowo niezależna, tworząc aktywny heterodimer HIF1.
Czynnik HIF1 odpowiedzialny jest za kontrolę ekspresji wielu genów kodujących białka, m.in. GLUT 1, 3, 4, VEGF, NFkB, Bcl-2. Po połączeniu aktywnego heterodimera HIF1 z kofaktorami CRB/p300 następuje rozpoznanie zlokalizowanych w obrębie regionów promotorowych genów sekwencji HRE (ang. Hypoxia response elements) oraz regulacja ekspresji znajdujących się pod kontrolą HIF1 genów. Odnotowano dodatnią korelację między ekspresją HIF1i GLUT1 i 3. Aktywacja HIF1 wiąże się z zahamowaniem oddychania mitochondrialnego i zintensyfikowaniem procesu glikolizy, co jest wynikiem wzmożonej ekspresji mitochondrialnej kinazy dehydrogenazy pirogronianowej, inaktywującej dehydrogenazę pirogronianową [10,11,12].
Zmniejszona dyfuzja tlenu, substancji odżywczych prowadzi do próby adaptacji komórek nowotworowych do tych warunków, poprzez metaboliczne przeprogramowanie i zwiększoną ekspresję czynników proangiogennych. Konsekwencją tego jest nasilenie angiogenezy nowotworowej. Jednak za szybki przyrost masy guza i niewielka wydajność nowopowstałych naczyń krwionośnych prowadzi do wystąpienia ognisk nekrozy poprzez hipoksję znacznych obszarów tkanki nowotworowo zmienionej. Jest to tzw. przykład „błędnego koła”, pokazany na rycinie 1 [3].
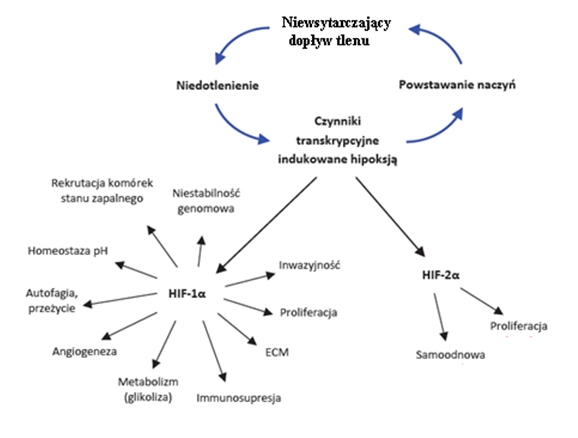
Ryc 1. Schemat „błędnego koła”: powiązanie powstawania nowych naczyń krwionośnych w obrębie guza z hipoksją [3].
Podsumowanie
Proces uzyskiwania energii przez komórki zmienione w wyniku procesu transformacji nowotworowej ma charakter złożony. Ponadto jest cały czas badany, poprzez co wiedza na ten temat jest pogłębiana. Zaowocowało to wysunięciem nowych teorii odnośnie metabolizmu nowotworu oraz sugestią praktycznego wykorzystania pozyskanych informacji w diagnostyce i leczeniu pacjentów onkologicznych. W związku z czym zasadne wydaje się dalsze przeprowadzanie badań w tym temacie [przyp. autora].
Literatura:
1. Ścibor – Bentkowska D, Czechot H. Komórki nowotworowe a stres oksydacyjny. Post. Hig Med. Dośw. (online) 2009; 63:58-72.
2. Szala S. Komórki mikrośrodowisku nowotworowego: cel terapii przeciwnowotworowej. Nowotwory 2007; 57(6): 633-645.
3. Szala S, Jarosz M. Nowotworowe naczynia krwionośne. Post Hig Med. Dośw.(online) 2011; 65:437-446.
4. Kozłowska J, Łamczańska I. Niestabiloność genetyczna – jej znaczenie w procesie powstawania nowotworów oraz diagnostyka diagnostyka laboratoryjna. Nowotwory 2010; 60 (6):548-553.
5. Dudziak K, Regulska- IIow B. Znaczenie ładunku glikemicznego diety w rozwoju chorób nowotworowych. Post. Hig. Med. Dośw. (online). 2013; 67:449-462.
6. Kidd J.G., Winzler R.J., Burk D.: Comparative glycolytic and re¬spiratory metabolism of homologous normal, benign, and malignant rabbit tissues. Cancer Res., 1944; 4: 547-553.
7. John A.P.: Dysfunctional mitochondria, not oxygen insufficien¬cy, cause cancer cells to produce inordinate amounts of lactic acid: the impact of this on the treatment of cancer. Med. Hypotheses, 2001; 57: 429-431.
8. Gasińska A, Janecka A, Adamczyk A, Słonina. Jak oddychają komórki nowotworowe ? Nowotwory 2013; 63 (2):124-131.
9. Jóźwiak P, Lipńska A. Rola transportera glukozy (GLUT1) w diagnostyce i terapii nowotworów. Post. Hig. Med. Dośw. (online) 2012; 66:165-174.
10. Dudziak K, Regulska- IIow B. Znaczenie ładunku glikemicznego diety w rozwoju chorób nowotworowych. Post. Hig. Med. Dośw. (online). 2013; 67:449-462.
11. Ke Q, Costa M. Hypoxia – inducible Factor – 1 (HIF – 1). Mol. Pharmacol. 2006; 70:1469-1480.
12. Menrad H, Werno C, Schmid T, Copanaki E, Deller T, Dehne E, Brüne B. Roles of Hypoxia – Inducible Factor 1α (HIF-1α) versus HIF-2α in the survival of hepatocellular tumor spheroids. Hepatology 2010; 51(6):2183–2192.