
Często odbierając wyniki sekwencjonowania konkretnego genu zastanawiacie się co dalej. Oczywiście można wrzucić ją do bazy danych (np. GenBank) albo dodać do publikacji, ale to jest najprostsze rozwiązanie. Przy użyciu narzędzi bioinformatycznych można zrobić dużo więcej.
1. Adnotacja sekwencji nukleotydowej i aminokwasowej.

Zbiór podstawowych informacji o sekwencji: długość, organizm, z którego pochodzi, potencjalna funkcja w oparciu o podobieństwo do sekwencji dostępnych w bazach danych. Podobieństwo w kontekście sekwencji homologicznych, rejony konserwowane i ważne pod względem pełnionej funkcji.
2. Identyfikacja motywów i domen.

Na podstawie analizy sekwencji pierwszorzędowej możliwa jest identyfikacja charakterystycznych motywów czy konserwowanych domen w białku (np. domeny funkcjonalne, motywy wiążące DNA/RNA, place cynkowe, itd.).
3. Lokalizacja potencjalnych helis transbłonowych.

Analizując sekwencje warto sprawdzić czy występują w niej helisy transbłonowe o charakterystycznym składzie aminokwasowym. Może mamy do czynienia z białkiem budującym kanał transbłonowy.
4. Identyfikacja elementów struktury drugorzędowej.

Lokalizacja elementów struktury drugorzędowej (alfa-helis i beta-wstęg) jest podstawową analizą na poziomie sekwencji i daje ona podstawowy zbiór informacji.
5. Położenia regionów nieuporządkowanych.
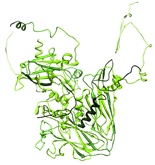
Olbrzymia część białek zawiera regiony, które są nieuporządkowane. Regiony nieuporządkowane pozbawione są stabilnej struktury trzeciorzędowej w warunkach natywnych. Funkcja fragmentów nieuporządkowanych w białkach ludzkich łączona jest ze schorzeniami, takimi jak nowotwory, choroby układu krążenia, amyloidoza, choroby neurodegeneracyjne i cukrzyca.
6. Kontakt poszczególnych aminokwasów z rozpuszczalnikiem.
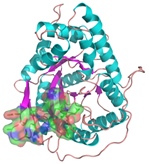
Identyfikując, które z aminokwasów są wystawione w stronę rozpuszczalnika, a które są zagrzebane w strukturze jesteśmy w stanie przewidywać funkcję regionów białka. Uzyskana informacja ma również wpływ na jakość modelowanej struktury przestrzennej białka.
7. Obecność potencjalnych oddziaływań białko-RNA/DNA lub białko-białko.
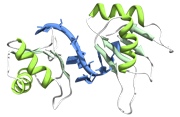
Często, aby uzyskać podstawowe informacje na temat biologii analizowanego systemu przewiduje się potencjalne oddziaływania białek z innymi białkami lub kwasami nukleinowymi.
8. Przewidywanie struktury trzeciorzędowej.
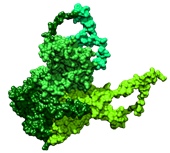
Przewidywanie struktury trzeciorzędowej umożliwia nam zidentyfikowanie zwoju białka oraz potencjalnych białek homologicznych, na podstawie których będzie można modelować strukturę przestrzenną badanego przez nas białka lub przewidzieć jego funkcję.
9. Budowa modeli przestrzennych białka.
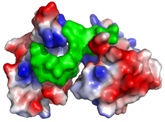
Dysponując zestawem powyższych informacji jesteśmy w stanie opracować model przestrzenny białka i zaproponować mechanizmy oddziaływania z innymi białkami, ligandami czy kwasami nukleinowymi.
10. Analizy filogenetyczne.
Analizy filogenetyczne to temat bardzo szeroki i wymagający zaawansowanych narzędzi, wiedzy i doświadczenia. Startując od jednej sekwencji jesteśmy w stanie zebrać kolekcję sekwencji homologicznych i przeprowadzić analizy filogenetyczne, które dadzą nam odpowiedź na pytania dotyczące pochodzenia sekwencji czy relacji w rodzinie sekwencji zbliżonych do siebie.
Tego typu dodatkowe analizy danych sekwencyjnych mogą wzbogacić wyniki doświadczalne oraz niejednokrotnie sugerują dalszą drogę projektowania eksperymentu. Otwarte podejście do stosowania narzędzi bioinformatycznych znacząco skraca czas planowania i przeprowadzenia eksperymentu oraz oszczędza pieniądze.
Autor artykułu:
Dr Anna Czerwoniec
VitaInSilica Sp. z o. o.
Krzemowa 1, Złotniki
62-002 Suchy Las
NIP: 9721237412
REGON: 301973876
www.vitainsilica.pl
office@vitainsilica.pl