PRZEDRUK, oryginał dostępny pod adresem www
Fragment skryptu: Biologia molekularna roślin
Uniwersytet Warszawski (www)
Instytut Biochemii i Biofizyki PAN (www)
Zakład Biologii Molekularnej Roślin (www)
Kierownik Zakładu: Prof. dr hab. Andrzej Jerzmanowski
Adres:
ul. Pawińskiego 5a,
02-106 Warszawa
Kontakt: tel. (+48 22) 592 5704,
E-mail: andyj@ibb.waw.pl
Zakład Biologii Molekularnej Roślin
Problematyka badawcza: Rola struktury chromatyny w regulacji rozwoju roślin oraz w odpowiedzi na czynniki stresowe i hormonalne. Prowadzone aktualnie badania mają na celu poznanie funkcji roślinnych kompleksów remodelujących chromatynę, histonu H1 i modyfikacji potranslacyjnych histonów rdzeniowych, a także opisanie proteomu jądrowego rośliny modelowej Arabidopsis thaliana.
Stosowane techniki: Większość metod biologii molekularnej, metody biochemii białek, analiza proteomiczna (mass-spec) i transkryptomiczna (mikromacierze), genetyka Arabidopsis thaliana (konstrukcja i analiza mutantów), metody bioinformatyczne.
_______________________________________________________________________________
RT-PCR jest techniką łączącą reakcję odwrotnej transkrypcji oraz reakcję PCR. Sprowadza się ona do amplifikacji specyficznego fragmentu RNA, dzięki czemu możliwa jest detekcja oraz oszacowanie poziomu ekspresji genów. Pod tym względem RT-PCR uzupełnia się, lub stosuje się zamiennie z takimi technikami biologii molekularnej jak Northern blot, hybrydyzacja In situ oraz mikromacierze DNA. Wśród zastosowań metody RT-PCR można wymienić m.in. badanie alternatywnych form splicingowych, wykrywanie markerów nowotworowych, detekcję transkrypcji genów wprowadzanych do organizmów transgenicznych oraz określanie wpływu mutacji na poziom ekspresji genów.
Pierwszym etapem RT-PCR jest reakcja odwrotnej transkrypcji, tj. synteza nici komplementarnego DNA (cDNA) na matrycy RNA, prowadzona przez enzym odwrotną transkryptazę. Właściwa reakcja PCR następuje dopiero po reakcji odwrotnej transkrypcji. Jest to konieczne z uwagi na to, że RNA nie jest matrycą dla termo stabilnych polimeraz DNA używanych w reakcji PCR.
Matrycowy RNA
Jako matrycowy RNA może służyć zarówno mRNA, jak i całkowity RNA komórkowy (ten wariant zostanie użyty podczas ćwiczeń). Aby osiągnąć dobrą wydajność odwrotnej transkrypcji, wyizolowany RNA musi być czysty – przede wszystkim nie powinien być zanieczyszczony DNA. Ewentualną obecność DNA w izolacji można sprawdzić w różny sposób, m.in. ustawiając reakcję kontrolną bez odwrotnej transkryptazy (w ćwiczeniu oznaczona „-RT”) lub amplifikując sekwencję zawierającą intron, który podczas dojrzewania mRNA jest wycinany.
O czystości próbki wyizolowanego RNA świadczy także stosunek absorbancji przy długościach fali 230, 260 oraz 280 nm. Dla czystego RNA stosunki absorbancji A260/A280, oraz A230/A260 wynoszą około 2 i ulegają zmniejszeniu lub podwyższeniu w obecności zanieczyszczeń (fenol, białka). Jakość (np. czy wystąpiła degradacja) oraz ilość RNA można ocenić na podstawie elektroforezy w żelu agarozowym.
Reakcja odwrotnej transkrypcji
Reakcję odwrotnej transkrypcji prowadzi się najczęściej przy użyciu primera oligo(dT) (Rys. 11a), który jest komplementarny do fragmentu poliA na 3’-końcu cząsteczek mRNA. Zastosowanie primera oligo(dT) pozwala na uzyskanie puli cDNA odpowiadającej całemu mRNA znajdującemu się w komórce (poza nielicznymi wyjątkami – m.in. mRNA wariantów histonów, których transkrypcja zależna jest od syntezy DNA). W szczególnych przypadkach (np. brak poliA w analizowanym mRNA, analiza innych klas RNA) stosuje się przypadkowe primery heksamerowe (Rys. 11b), które dają jednakową reprezentację całego RNA komórkowego, lub też primer specyficzny do określonej sekwencji (Rys. 11c). Do oceny jakości uzyskanego cDNA służy reakcja kontrolna PCR z użyciem primerów specyficznych do genu eksprymowanego konstytutywnie, np. aktyny.
<img src=”http://e-biotechnologia.pl/obrazki/RT_PCR.JPG” ALIGN=”right” alt=”odwrotna transkrypcja” HSPACE=10 VSPACE=10/> Rys. 11 Schemat reakcji odwrotnej transkrypcji z użyciem różnych typów primerów.
Półilościowy RT-PCR
Terminem tym określa się metodę, w której porównuje się ilość dwóch lub więcej cząsteczek RNA z jednej lub większej ilości izolacji, przy czym ilość transkryptu szacuje się na podstawie ilości produktu PCR powstałego na matrycy cDNA.
Aby możliwe było szacowanie poziomu ekspresji, w porównywanych reakcjach PCR musi się znajdować ta sama ilość cDNA, co można stwierdzić dzięki wewnętrznej kontroli (PCR z primerami specyficznymi do genu eksprymowanego konstytutywnie, np. aktyny). Liczba cykli w półilościowym PCR musi być ustawiona tak, aby zachować liniowość przyrostu ilości produktów w kolejnych cyklach.
Opis doświadczenia
W poniższym doświadczeniu wykorzystana będzie linia homozygotyczna – mutant insercyjny T-DNA h1.3_1 otrzymany z bazy mutantów GenTrap. Natomiast mutant h1.3miR został uzyskany poprzez wprowadzenie do roślin o genotypie typu dzikiego (Col-0) transgenu kodującego sztuczne microRNA do sekwencji genu H1.3. Uzyskano linie heterozygotyczne h1.3miR.
Cel doświadczenia:
Sprawdzenie na jakim poziomie wyrażany jest gen H1.3 w mutantach Arabidopsis thaliana h1.3_1 i h1.3miR.
Schemat doświadczenia
Etap 1.
Genotypowanie metodą PCR roślin h1.3miR oraz Col-0 (jako kontrola) pod względem obecności transgenu kodującego oporność na herbicyd Basta.
a) izolacja DNA genomowego,
b) amplifikacja fragmentu DNA metodą PCR.
Etap 2.
Analiza poziomu ekspresji genu H1.3 za pomocą metody RT-PCR.
a) izolacja RNA,
b) odwrotna transkrypcja (RT),
c) półilościowy RT-PCR.
Dzień 1. Izolacja DNA genomowego na małą skalę, genotypowanie roślin
Materiały:
– liście roślin
– probówki 200 μl do PCR,
– końcówki do pipet (tipsy),
– termocykler.
Odczynniki:
– chelex 10%
– odczynniki do PCR (bufor i polimeraza Pfu, startery, MgCl2, dNTP)
Wykonanie:
1. Fragment liścia o wielkości wieczka od eppendorfa wrzucić do probówki PCR o pojemności 200 μl.
2. Dodać 50 μl 10% Chelex-u uprzednio silnie wymieszanego (vorteks przez 30 sekund).
3. Ucierać liść tipsem (o pojemności 200 μl) przez około 15 sekund, do pojawienia się zielonego zabarwienia roztworu chelexu.
4. Dodać kolejne 50 μl 10% Chelex-u uprzednio silnie wymieszanego (vorteks przez 30 sekund).
5. Inkubować w termocyklerze w 98ºC przez 20 min. i 2 min w 22ºC.
6. Próbki wirować przy maksymalnych obrotach przez 5 min. (w adapterach wykonanych z probówek eppendorfa 1,5 ml i 0,5 ml) w mikrowirówce.
7. Pobrać 20 μl supernatantu zawierającego DNA. Nie pobierać złoża ani fragmentów liści!
Tak otrzymane DNA można przechowywać w 4°C.
8. Przygotować 1% żel agarozowy.
9. Nanieść na 1% żel agarozowy 5 μl DNA + 0,5 μl obciążnika do DNA.
10. Przygotować mieszaninę na 3 reakcje PCR do określenia obecności genu BAR wg schematu: (próbki przygotowywać w lodzie).
Mieszanina na 1 reakcję PCR:
– Matryca DNA 2 μl
– Mix primerów BAR 5 μl,
– Bufor Pfu (10x) 5 μl,
– dNTP (10mM) 8 μl,
– MgCl2 (25mM) 6 μl,
– Polimeraza Pfu 1 μl,
– H2O milliQ do 50 μl,
Warunki reakcji PCR:
– hold 94ºC,
– 94ºC 2 min.
– 94ºC 30 s
– 60ºC 30 s 35 cykli
– 72ºC 45 s
– 72ºC 3 min.
– hold 4ºC
11. Po zakończeniu programu dodać 5 μl obciążnika.
12. Nanieść na 1% żel agarozowy 20 μl reakcji.
Dzień 2 i 3. Izolacja całkowitego RNA
Należy pamiętać o tym, że niepowodzenie izolacji RNA zwykle wynika z zanieczyszczenia próbki RNazami (głównie z naskórka rąk), powodującymi degradację RNA.
Materiały:
– liście roślin
– moździerze
– tipsy (wolne od RNaz)
– rękawiczki (wolne od RNaz)
– vorteks pod wyciągiem
– wirówka pod wyciągiem
Odczynniki:
– bufor do homogenizacji: 100 mM Tris-HCl pH 8, 5 mM EDTA, 100 mM NaCl, 0,5% SDS, 2-merkaptoetanol,
– kwaśny fenol pH 4.0
– mieszanina fenol/chloroform/alkohol izoamylowy (24:24:1) pH 4.0
– chloroform
– H2O milliQ (wolna od RNaz)
– 3M octan sodu pH 5.2 (4ºC)
– izopropanol (-20ºC)
– etanol 70% (-20ºC)
Wykonanie: (Należy pracować w rękawiczkach!)
1. Przygotować bufor do ekstrakcji. Pod wyciągiem na każde 500 μl buforu homogenizacyjnego dodać 5 μl 2-merkaptoetanolu.
2. Zebrać równe ilości materiału roślinnego (roślina kontrolna Col-0 oraz mutanty h1.3_1 i h1.3miR).
3. Utrzeć tkankę w ciekłym azocie w moździerzu.
4. Utartą tkankę przenieść do probówki eppendorfa 1,5 ml schłodzoną szpatułką i zalać 500 μl buforu ekstrakcyjnego.
5. Wytrząsać (vorteks) przez 30 sekund.
Pod wyciągiem (w fartuchu, okularach i rękawiczkach)
6. Dodać 250 μl kwaśnego fenolu (uwaga substancja szkodliwa!).
7. Wytrząsać 30-60 sekund.
8. Dodać 250 μl chloroformu (uwaga substancja szkodliwa!).
9. Wytrząsać 30-60 sekund.
10. Zwirować (w mikrowirówce pod wyciągiem).
10 min. przy maksymalnej prędkości.
11. Przenieść fazę wodną (górna warstwa ok. 500 – 600 μl) do nowej probówki eppendorfa.
12. Dodać 600 μl mieszaniny fenol/chloroform/ alkohol izoamylowy – warstwa dolna (uwaga substancja szkodliwa!).
13. Wytrząsać 30 sekund.
14. Zwirować (pod wyciągiem) 10 min. przy maksymalnej prędkości.
15. Przenieść warstwę wodną (górna warstwa 400 – 500 μl) do nowej probówki.
Od tego etapu pracować szczególnie ostrożnie, aby zapobiec zanieczyszczeniu próbek RNazami (rękawiczki).Próbki trzymać na lodzie
16. Dodać 50 μl 3M octanu sodu pH5,2 (0.1v/v).
17. Dodać 450 μl (1:1 v/v) zimnego (-20ºC) izopropanolu, zamieszać.
18. Inkubować 20 minut w -20ºC.
19. Zwirować 30 min. przy maksymalnych obrotach.
20. Delikatnie usunąć supernatant.
21. Suszyć osad przez 10 min. w temp. pokojowej.
22. Zawiesić w 500 μl wody wolnej od RNaz.
23. Dodać 500 μl 4M LiCl (końcowe stężenie 2M).
24. Inkubować w 4ºC przez noc.
25. Zwirować 30 min. przy maksymalnych obrotach, bardzo delikatnie usunąć supernatant (słabo widoczny osad RNA może się odkleić).
26. Przemyć osad 1 ml zimnego (-20ºC) 70% etanolu.
27. Zwirować 5 min. przy maksymalnych obrotach, bardzo delikatnie usunąć supernatant (słabo widoczny osad RNA może się odkleić).
28. Osad zwirować powtórnie 10 sekund i bardzo delikatnie odciągnąć resztkę etanolu.
29. Powtórzyć czynność z poprzedniego punktu.
30. Suszyć osad przez 10 min. w temp. pokojowej.
31. Zawiesić osad w 30 μl wody.
32. Inkubować RNA 2 min. w 65ºC, worteksowaćc30 sekund, schłodzić w lodzie. 33. Określenia ilości i czystości wyizolowanego RNA przy użyciu spektrofotometru NanoDrop lub innego.
34. Przygotować 1,2% żel agarozowy.
35. Zmieszać 5 μl RNA z 5 μl obciążnika do RNA, denaturować 10 min. w 70°C.
36. Nanieść preparat na żel agarozowy i przeprowadzić elektroforezę przy napięciu 90 V, obejrzeć żel i zrobić zdjęcie.
Dzień 4. Synteza jednoniciowego cDNA za pomocą RevertAid First Strand Synthesis Kit (Fermentas).
1. Przygotować mieszaninę reakcyjną (na lodzie, w probówkach do PCR o pojemności 500 μl) Objętość użytego do reakcji preparatu RNA uzależniona jest od stężenia i jakości wyizolowanego RNA oraz poziomu ekspresji danego genu.
– 1 μg RNA z rośliny kontrolnej Col-0 (probówka 1)
– 1 μg RNA z rośliny kontrolnej Col-0 do reakcji -RT (probówka 2, -RT)
– 1 μg RNA z rośliny h1.3miR (probówka 3)
– 1 μg RNA z rośliny h1.3miR do rekcji -RT (probówka 4, -RT)
– 1 μg RNA z rośliny h1.3_1 (probówka 5)
– 1 μg RNA z rośliny h1.3_1 do rekcji -RT (probówka 6, -RT)
Do probówek 1 – 6 dodać:
– primer oligo(dT) 0,5 μg/μl – 1 μl
– woda sterylna (Milli-Q) dopełnić do 13 μl
2. Mieszaninę inkubować w termocyklerze w 70°C przez 5 min., a następnie probówkę umieścić w lodzie. Inkubacja w 70°C pozwala na denaturację drugorzędowych struktur mRNA.
3. Do zdenaturowanego RNA dodać 7 μl mieszaniny RT+ lub RT zawierającej:
– 5x stężony bufor reakcyjny
– 10mM mix dNTP
– odwrotną transkryptazę (+RT) lub wodę (- RT).
4. Inkubować w 42°C przez 1 h. W tym etapie zachodzi reakcja odwrotnej transkrypcji. Reakcja jest zatrzymywana przez ogrzanie mieszaniny do 70°C przez 10 min.
cDNA należy przechowywać w -20°C. Zsyntetyzowane cDNA posłuży jako matryca do amplifikacji analizowanych sekwencji metodą PCR.
Dzień 5. Półilościowy multiplex PCR
Multiplex PCR polega na amplifikacji jednocześnie kilku sekwencji w jednej reakcji PCR. Warunkiem powodzenia takiego typu reakcji jest optymalizacja jej warunków – odpowiedni dobór buforu oraz primerów, liczby cykli, jednakowej temperatury przyłączenia starterów, itd. Multiplex PCR ma istotną przewagę nad zwykłym PCR. W półilościowym multiplex RT-PCR produkty amplifikacji określonych transkryptów oraz kontroli uzyskane są w jednej reakcji i w jednakowych warunkach, co pozwala na lepsze porównanie ich ilości.
Dla trzech wariantów eksperymentalnych (Col-0, h1.3_1 i h1-3miR):
1. Przygotować mieszaninę do PCR według schematu (próbki przygotowywać w lodzie).
Mieszanina na 1 reakcję:
– Matryca cDNA 1-2 μl
– Mix primerów (M) 4 μl
– Bufor Pfu (10x) 5 μl
– dNTP (2,5mM każdy) 8 μl
– MgCl2 (25mM) 6 μl
– Polimeraza Pfu 1 μl
– H2O milliQ do 50 μl
Warunki reakcji PCR:
– hold 94ºC
– 94ºC 2 min.
– 94ºC 30 s
– 68ºC 6 min. 27 cykli
– hold 4ºC
2. Po zakończeniu reakcji PCR do probówek należy dodać 1/10 objętości barwnika do elektroforezy.
3. 20 μl próbki nanieść na 1,2 % żel agarozowy.
Elektroforeza DNA w żelu agarozowym
Materiały
– agaroza
– bufor TBE (45 mM Tris-boran, 1 mM EDTA)
– bromek etydyny
– obciążnik DNA (DNA loader)
– standard wielkości (marker)
Wykonanie
1. Odważyć 0,5 g/0,6 g agarozy (1% / 1,2% )
2. Agarozę przesypać do kolby, dodać 50 ml buforu TBE
3. Rozpuścić agarozę przez podgrzanie w kuchence mikrofalowej
4. Schłodzić kolbę pod bieżącą wodą, dodać bromku etydyny do stężenia 0,5 μg/ml.
5. Wlać żel do wanienki, włożyć grzebień, pozostawić do zastygnięcia
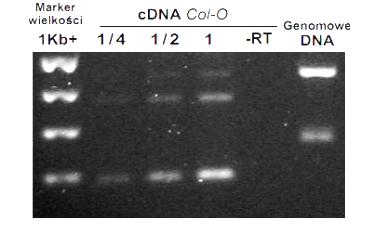
6. Prowadzić elektroforezę w buforze TBE przy napięciu ok. 100 V w obecności markera wielkości.
7. Żel obejrzeć i sfotografować na trans iluminatorze UV z kamerą. Oczekiwany wynik dla roślin dzikich z uwzględnieniem kontroli wewnętrznej (aktyna) przedstawia Rys. 12 Rys. 12. Rozdział elektroforetyczny produktów amplifikacji histonu H1.3 i aktyny.